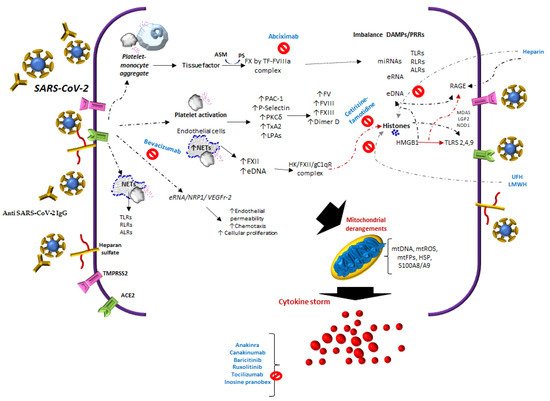
During COVID-19 infection, SARS-Cov-2 interacts with Angiotensin-Converting Enzyme 2 (ACE2)E2, NRP1, endothelial cells, platelets, neutrophil extracellular traps (NETs)NETs, thrombin, extracellular DNA (eDNA)DNA, and histones, inducing heterogeneous clinical manifestations characterized by endothelial damage, microthrombosis, and inflammation.
1. Introduction
COVID-19 shows heterogeneous clinical expression, which is associated with thrombosis and microangiopathy. There are various opposing theories related to the virus. On the one hand, it could be associated with intravascular coagulation
[1][5]. On the other hand, it could correspond to complement-mediated thrombotic microangiopathies
[2][3][6,7]. Hypercoagulation
[4][8], platelet hyperactivity
[5][9], and abnormal fibrinolysis
[6][7][10,11] might explain the diversity of macrovascular and microvascular thrombosis expression, depending on the study method. In the selection criteria for study subjects and the general population, the frequency of thrombosis is variable. For example, results from autopsies report that in 87% of microthrombosis cases
[8][12], coagulopathy is found in up to 50% of fatalities
[9][13], and macrothrombosis, such as deep vein thrombosis and pulmonary thromboembolism, is found in up to 40%
[10][11][14,15].
Several signaling routes have been reported to play a role in the mechanisms of immunothrombosis or thrombo-inflammation
[12][13][16,17] and cytokine storms in COVID-19 (
Figure 1). The sequence of these events in SARS-CoV-2 infection is related to the interactions of different cells and molecules, such as Angiotensin-Converting Enzyme 2 (ACE2), tissue thromboplastin or tissue factor (TF), neutrophil extracellular traps (NETs), extracellular DNA (eDNA) and RNA, histones
[14][18], anti-PF4/heparin IgG antibodies, antiphospholipid antibodies, neutrophil-platelet aggregates, and monocyte-platelet, among others.
Figure 1. The reported studies of COVID-19 infer that there are multiple activation or inhibition routes in platelets and endothelium, this involves the release of tissue thromboplastin (TF), with the participation of monocyte-platelet, neutrophil-platelet aggregates, complement-TF-NETs, and histones. In addition, to immune complexes SARS-CoV-2 spike/anti-spike IgG, anti-PF4/heparin IgG antibodies and antiphospholipid antibodies.
1.1. Factors of the Contact System
In COVID-19, specifically, the expression of FXIIa increases in lung tissue. In addition, this factor is colocalized with NETs in the lungs, indicating that the accumulation of NETs leads to greater activation of FXII due to a defect in the clearance of NETs by DNases, contributing to procoagulant activity [15][22]. This is also related to the activity of FXII in the blood coagulation system and increases in DNA and H4 histones [16][23]. Histones contribute to microvascular thrombosis and competitively inhibit plasmin to delay fibrinolysis [17][24].
1.2. Tissue Factor
In COVID-19, increased circulating extracellular vesicle TF activity has been reported, which correlates with the markers of thrombosis such as D-dimer [18][32]. It is important to point out that TF expression may be inhibited by platelet P-selectin (CD62P) neutralization or integrin αIIb/β3 blocking, as with abciximab [19][31].
1.3. Neutrophil Extracellular Traps and Molecule Release
In COVID-19, the platelet/NETs/TF/thrombin axis is enhanced by complement activation [20][33]. Not only do neutrophils release eDNA, but also macrophages, eosinophils, and mast cells. These appear at different stages of thrombosis [21][22][37,38], and similarly in tumor cells [23][34].
In COVID-19 patients, platelet activation products, such as TXB2 and proteins from platelet α-granules PF4/CXCL4 and PDGF, are also released and found in tracheal aspirates [19][31].
Of the molecules with NETs that are released, it has been suggested that free DNA may be the cause of a more severe pathology in COVID-19 [24][39]. Moreover, the manifestations of severity could be related not only to eDNA, but also to other alarmins, such as extracellular heat-shock proteins and HMGB1, mentioned above, in addition to diverse self-nucleic acids, including nuclear DNA, ribosomal RNA, extracellular RNA (eRNA), micro-RNAs, and histones [25][40], Figure 1.
In COVID-19, eDNA from NETs and histones could also explain thrombosis in severe forms [26][45]. The half-life of eDNA is around 4–30 min [27][46]. Its clearance is regulated by different factors, such as 1) Serine proteases, e.g., Factor VII activating protease; cysteine proteases, e.g., caspase-activated DNAse [28][47]; DNASE1; and deoxyribonuclease 1-like 3 (DNASE1L3) [29][48].
A crucial point of NETs is the equilibrium or balance between the release of eDNA and eRNA and their respective nucleases, which are required to maintain homeostasis. In COVID-19, a significant increase in NETs is observed, and therefore an excess of eDNA. In clinical trials, the human DNase I enzyme, (Dornase Alpha) is being evaluated to reduce the severe symptoms of COVID-19
[30][56].
1.4. Platelets-SARS-CoV-2/Angiotensin-Converting Enzyme 2
Thrombocytopenia in COVID-19 is an indicator of poor prognosis, particularly when it decreases in the first 7 days after admission to the hospital. Thrombocytopenia is an independent risk factor associated with in-hospital mortality. Liu et al. [31][57] found that an increase of about 50 × 109 /L over the whole range of platelet, decreases mortality. Adding support to the means of platelet activation, several studies show that platelets are activated in COVID-19 patients [32][58], i.e., there is an increase in young immature platelets named reticulated platelets (RPs). These are associated with high platelet turnover and arterial thrombotic events. In COVID-19, PRs or the immature platelet fraction (IPF) are similar to patients with acute myocardial infarction [33][59].
1.5. Heparin-Induced Thrombocytopenia
Considering that one of the main characteristics of COVID-19 is hypercoagulability, and therefore the increased risk of venous and arterial thrombosis, it is necessary to differentiate from Heparin-induced thrombocytopenia (HIT)
[34][35][36][67,68,69], particularly secondary to the use of vaccines
[37][70]. Uaprasert et al.
[38][71], in a systematic review and meta-analysis, found a pooled incidence of HIT of 0.8%, being slightly higher in critically ill COVID-19 patients.
2. Cytokine Storm Syndrome
During a COVID-19 infection, immune cells flood the lungs and attack them instead of protecting them. The imbalance between PAMPs and pattern recognition receptors (PRRs) could fall into cytokine storm syndrome (CSS) or a specific syndrome from the family of conditions characterized by a cytokine storm such as macrophage activation syndrome. This is associated with autoimmune disorders, hemophagocytic lymphohistiocytosis genetic or secondary to different disorders, and cytokine release syndrome
[39][87]. CSS is also associated with delayed secretion of type I and III interferons
[40][88], and low levels thereof
[41][89], probably due to membrane protein in SARS-CoV-2 via RIG-I/MDA-5-MAVS signaling
[42][90]. Furthermore, CSS has hypercytokinemia
[43][91], macrophage polarization from M2 to M1
[44][92], with activation of the Plcγ2 pathway and a reduction in Tmem178 levels in macrophages
[45][93], T-cell cytotoxicity defects
[12][16], complement activation
[46][94], and increased NETs
[47][48][95,96]. As a result of all these changes, patients suffer from hyperinflammation
[49][97], cytokine release
[50][98], cytokine storms
[51][52][99,100], multiorgan disease
[53][101], and thrombosis
[11][15]. Therefore, various inhibitors have been suggested
[54][102], several of which are being used in clinical trials. Some preliminary studies report a significant reduction in mortality, by Anakinra
[55][103] and Canakinumab
[56][104] for IL-1 inhibition, and baricitinib
[57][105] and ruxolitinib
[58][106] for JAK inhibition. However, others have divergent findings, e.g., tocilizumab
[59][60][107,108] and sarilumab
[61][109] for IL-6 inhibition (
Figure 1). Moreover, molecules with immunomodulatory and antiviral properties that modulate cytokines and interferons in immunosuppressed subjects such as inosine pranobex
[62][63][110,111] have been used in clinical trials with promising results
[64][112].
3. Influence of Heparanase, Heparin and Heparinoids in Complications from COVID-19
Heparanase (HPSE) is an endo-β-D-glucuronidase that has specificity for HS and heparin polysaccharide chains. It participates in the metabolism of HS in the extracellular matrix and its activity is modified in inflammation, cancer and cell migration
[65][113]. Acting as a cofactor of TF, HPSE increases the activity of FX and interacts with the TF pathway inhibitor, acting as a procoagulant
[66][114]. It also has platelet hyperactivity and thrombotic activity
[67][115]. HPSE is increased in COVID-19 patients and is related to pathogenicity
[68][116]. HPSE interacts with other molecules such as RNA, causing vascular leakage and inflammation. Low-molecular-weight heparins (LMWH) are potent inhibitors of HPSE, thrombus, and inflammation
[53][69][101,117], and they also neutralize histones
[70][118].
Histone levels together with HPSE levels may explain interindividual sensitivities to heparin (unfractionated heparin (UFH) and LMWH) or heparinoids in COVID-19 patients
[71][72][119,120]. This means that the heparin used to treat microthrombosis in these subjects also participates in the inhibition of histones and could decrease its toxicity. The effect of LMWH is not always sufficient, as mentioned above, or it could be reversed
[73][121]. In COVID-19 patients, the use of LMWH is very important due to its anticoagulant, antiviral, and anti-inflammatory effects
[74][122]. However, the need for higher doses of LMWH has been observed in critically ill patients
[75][123], and the use of oral anticoagulants is required, namely dabigatran, apixaban and rivaroxaban
[76][77][124,125].
Heparin/ heparan sulphate competes with SARS-CoV-2 and reduces its entry into the body
[78][79][126,127], because, in the SARSCoV-2 spike (S) protein, the receptor-binding domain (RBD) in the S1 subunit has an ectodomain that interacts with heparan sulfate (HS)
[80][128], and the RBD region in S protein in SARS-CoV-2 interacts with 2-O or 6-O sulphate groups of heparin or enoxaparin
[81][129]. Additionally, it has been reported that heparan sulphate inhibitors, such as mitoxantrone, sunitinib, and BNTX, could block entry of the virus
[82][130]. In general, high negatively charged proteins, such as heparin, protein C, and pentraxin, neutralize histones
[83][131]. Heparin has been shown to act directly against circulating histones, but its action does not depend on its anticoagulant function
[84][132].
Among the heparin derivatives that have been proposed for the treatment of COVID-19 are heparinoids. Sulodexide is a heparinoid containing 80% iduronyl glycosaminoglycan sulphate (IGS) or fast-moving heparin and 20% dermatan sulphate (DS). IGS interacts with and increases antithrombin and heparin cofactor II (HCII)
[85][133], while DS interacts with HCII. This combination has properties resembling those of UFH
[86][134]. Sulodexide also has profibrinolytic activity, which reduces the neo-synthesis of proinflammatory cytokines and inhibits histones
[87][135]. It releases an inhibitor of the endothelial TF pathway, inhibiting FVIIa and FXa
[88][89][136,137]. The prolonged use of sulodexide produces a “release and depletion” effect as observed with UFH
[90][138]. Nevertheless, the use of heparin also has some limitations, such as its inability to inactivate antithrombin-heparin complex when it is bound to fibrin. Dermatan sulphate-HCII complex has been reported to inactivate fibrin-bound thrombin
[91][139]. The RBD region in the S protein in SARS-CoV-2 interacts with 2-O or 6-O sulphate groups of heparin or enoxaparin
[81][129]. Therefore, heparin, and most likely heparinoids, inhibit cellular interaction with the virus.
It is evident that, in addition to the factors related to the pathogenesis of SARS-CoV-2, other pathogenic factors are associated with thrombosis and inflammation, such as HPSE, eDNA, eRNA, micro-RNAs, and histones. Many of these molecules are being studied in order to find drugs to treat COVID-19.