 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Monica Fedele | + 2151 word(s) | 2151 | 2021-10-26 08:29:56 | | | |
| 2 | Camila Xu | Meta information modification | 2151 | 2021-11-18 02:21:03 | | | | |
| 3 | Camila Xu | Meta information modification | 2151 | 2021-11-18 09:06:32 | | | | |
| 4 | Monica Fedele | Meta information modification | 2151 | 2021-11-18 10:52:48 | | |
Video Upload Options
Reprogramming of metabolism is now recognized a hallmark of carcinogenesis as metabolic changes, such as those related to glucose, glutamine and lipids, are tightly related to the proliferation, invasion, migration, radiosensitivity, and chemosensitivity of several tumors, including thyroid cancer.
1. Introduction
Thyroid cancer (TC) represents the most common endocrine malignancy all over the world, with a steady increase in both the incidence and the mortality rate for the more aggressive forms [1]. According to the most recent epidemiologic studies in United States, TC incidence increased, on average, 3.6% per year during the period 1974–2013, mainly due to an increase in the incidence of papillary thyroid carcinoma (PTC) [1], and it has been estimated that by 2030 TC will be the fourth leading cancer diagnosis in the United States [2]. Accordingly, a recent deep analysis of the Global Burden of Disease 2019 database has calculated that the global incidence of TC has continued to increase in the past three decades [3]. Some of the highest TC incidence worldwide has been reported in Italy where, under the age of 45, TC was the second most common cancer among women (after breast cancer), and the fifth most common among men [4]. The most frequent TC (84% of all TC) is PTC, a differentiated TC (DTC) deriving from epithelial follicular cells. It is generally characterized by an indolent growth and a good prognosis after adjuvant radioiodine (RAI) treatment; the 5-year relative survival rate for patients who had TC diagnosed during the period 2008–2014 was 98%, and it refers mainly to the most prevalent PTC [5]. However, 20–30% of PTC cases show a more aggressive behavior and patients experience relapse/persistence and/or development of lymph node and visceral metastases with consequent increased mortality, despite the use of targeted therapeutic options, such as tyrosine kinase inhibitors (TKI), including sorafenib and lenvatinib [6][7]. During 1994–2013, incidence-based mortality increased 2.9% per year for advanced-stage PTC [1]. Due to the high global incidence of PTCs, the percentage of those RAI-resistant (RAI-R) has a significant impact and it is therefore imperative to find new therapeutic strategies. The aim of our review is to analyze the possibility that the intercross between epithelial-to-mesenchymal transition (EMT) and metabolism could be exploited to find such strategies. These aggressive forms of PTC exhibit loss of differentiation characteristics, including loss of sodium iodine symporter expression/function, resulting in RAI treatment failure and high mortality. At the molecular level, this loss of differentiation is related to the degree of activation of the mitogen-activated protein kinase (MAPK), which is highest in tumors with BRAF mutations [8].
On the other side, anaplastic thyroid carcinoma (ATC), the most undifferentiated TC, is a rare but devastating disease. It accounts for only 2–5% of all TC cases and is associated with a median overall survival (OS), greatly improved in the last years thanks to the targeted therapy, of 15.7 months, a median 1-year survival of 59%, and a median 2-year survival of 42%, despite aggressive multimodal management [9][10][11]. Current management of ATC consists primarily of surgical resection, combined with adjuvant chemoradiation followed by targeted therapy (dabrafenib and trametinib therapy in patients harboring the BRAF V600E mutation) [12]. The pathogenesis of ATC is still debated. Most studies support a gradual dedifferentiation from DTC to poorly differentiated thyroid carcinoma (PDTC), and eventually to ATC, with the progressive accumulation of somatic pro-cancer mutations. This is supported by the fact that 18–37% of ATC cases result from longstanding goiters or DTC lesions, where ATC occurs concurrently in 30–89% of cases, and ATC sometimes develops following treatment failure of DTC and PDTC. Genomic analyses have further demonstrated shared mutations between co-existing ATC and DTC or PDTC lesions, suggesting a common parent cell [13]. Another theory states that ATC could arise from cancer stem cells (CSCs) that are derived from adult stem cells present within a thyroid niche having accumulated genetic mutations that drive the tumor development [14]. For both theories, EMT plays a pivotal role. In fact, a DTC could lead to ATC as a result of either a dedifferentiation process or the development of CSCs, and both depend on EMT. CSCs are in turn the main responsible of cancer resistance [15], and therefore EMT is a cellular process associated with both tumor progression and TC resistance to therapy. Hence, understanding the biology of EMT and the reverse mesenchymal-to-epithelial transition (MET) process should lead to the design of more effective drugs to target cancer cells, including CSCs.
2. The Warburg and Reverse Warburg Effects
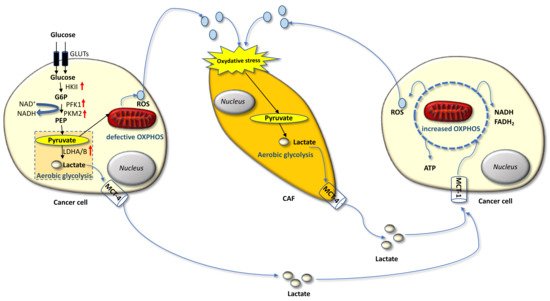
Carcinoma cells show preferential use of lactate-generating glycolysis over the more energy-efficient route of oxidative phosphorylation (OXPHOS), which produces more ATP per glucose molecule than glycolysis [16][17][18][19][20]. This altered metabolism, named “Warburg effect”, implies that cancer cells have increased glucose uptake and lactate secretion, and allows cancer cells to gain an advantage in terms of growth and survival, possibly due to increased carbon utilization, hypoxic adaptation, and increased rate of ATP production. More recently, similar metabolic changes have been described in cancer-associated fibroblasts (CAFs) present in the tumor microenvironment (TME), often as a result of oxidative stress induced by hydrogen peroxide secreted by cancer cells. CAFs in turn increase their own production of reactive oxygen species (ROS), resulting in the induction of aerobic glycolysis and consequent production and secretion of lactate and pyruvate. These metabolites are transferred to cancer cells via inflammation, where they are metabolized into mitochondria to generate new ATP, which assists tumor progression. This metabolic interplay between different tumor cell compartments is called “reverse Warburg effect” and facilitates cancer cell anabolism through catabolic reactions pursued by the TME [21][22][23][24][25]. The reverse Warburg effect can occur not only between CAFs and tumor cells but also between different tumor cells, one of which being hypoxic and hypersecreting intermediate catabolites such as lactate and glutamine. Metabolic coupling with glycolysis occurring in some cancer cells and OXPHOS in other cancer cells promotes cell proliferation and survival. In this multi-compartment metabolism, a key role is played by the lactate monocarboxylate transporters MCT-1 and MCT-4, which mediate the influx into the cell and the efflux from the cell, respectively [26] (Figure 1).
3. Metabolic Reprogramming in Thyroid Cancer
Metabolic rewiring towards an enhanced glycolytic phenotype primarily involves increased glucose uptake and glycolysis flux, mitochondrial dysfunction, and a more acidic TME, playing a critical role in tumor aggressiveness. In other words, malignant tumor cells alter their glucose metabolism to enhance aerobic glycolysis so that they can maintain their metastatic potential.
Amino acids metabolism has a critical role in maintaining cellular metabolic homeostasis. Among all amino acids, glutamine has the greatest consumption during tumor progression and is considered the most important substrate of the cancer cells. It has an essential role in nucleotide and non-essential amino acids synthesis, as well as in providing substrates for the tricarboxylic acid (TCA) cycle, which fuels tumor growth [27]. In particular, TCA cycle is maintained by glutamic acid derived from the conversion of glutamine through the process of glutaminolysis. Consistently, glutamic acid has been found increased in the plasma of patients with thyroid nodules, consisting of 19 PTCs and 16 multinodular goiters, compared to 20 healthy controls [28]. In this pilot study, a panel of significantly altered metabolites, including some associated with amino acids metabolism, such as cysteine and cystine as well as glutamic acid, was identified by untargeted gas chromatography-mass spectrometry in the plasma of patients with PTC nodules compared to healthy subjects. Differently from glutamic acid, cysteine and cystine were decreased in PTC patients and their levels correlated with the tumor stage [28].
Conversely, in a previous study, cysteine and most amino acids were found significantly up-regulated in PTC tissue (collected from 57 patients) compared to adjacent non-tumor tissue [29]. Cysteine is a precursor for glutathione (GSH) biosynthesis, which plays an essential role in supporting intracellular redox homeostasis by extinguishing ROS from mitochondrial respiration. Cancer cells require exogenous cysteine for GSH synthesis to protect themselves from ROS in order to maintain cell proliferation and resistance to apoptosis [30]. Therefore, decreased plasma levels of cysteine and cystine in patients with thyroid nodules may be explained by the higher consumption of cysteine in the cancer cells. Consistently, in the study by Abooshahab and coworkers, significantly altered metabolites between PTC nodules and healthy persons were also associated with GSH biosynthesis. Overall, they found that the metabolism of about 11 amino acids, including metabolites related to GSH biosynthesis, but also methionine, glycine, serine, threonine, and phenylalanine, had been changed in plasma of patients with PTC nodules compared to healthy subjects. Moreover, the TCA cycle, fatty acids (FA), and purine and pyrimidine metabolism were significantly changed as well [28].
4. Thyroid Cancer Progression and Reciprocal Role of Epithelial-Mesenchymal Transition and Metabolic Rewiring
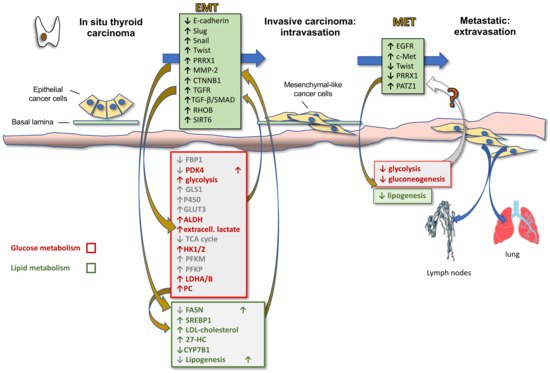
Activation of EMT, a process by which epithelial cancer cells acquire mesenchymal features, is a key determinant of cancer progression toward an invasive and metastatic phenotype. By acquiring mesenchymal features, cancer cells, in fact, lose cell-to-cell junctions and gain the capacity to migrate and invade the basal lamina thanks to a complex reprogramming of transcription through epigenetic changes. In TC progression, the tumor cells undergo EMT, becoming spindle shaped and invading tumor stroma. Molecular changes include reduced E-cadherin expression levels and increased expression of Snail, Slug, Twist, Paired Related Homeobox 1 (PRRX1), and other EMT-related genes. Hence, first intravasation into the blood and/or lymphatic vessels and then extravasation in distant metastatic sites, such as the lymph nodes and lungs, occur. After a variable time in the quiescence state, the tumor cells are subjected to MET to colonize distant organs forming secondary tumors (Figure 2). During this last phase there is a decrease in the expression of Twist and PRRX1 and an increase in the expression of epidermal growth factor (EGFR) and c-Met [15]. Indeed, well-differentiated TC and normal thyroid express high levels of E-cadherin, but do not commonly express Snail and Twist [31]. However, the leading front of PTCs, as well as ATCs, frequently express EMT markers, such as vimentin and Snail, Slug and Twist, but not E-cadherin [15][32][33].
During EMT cancer cells also acquire stem cell features that allow them to resist to different treatment options. Based on the CSC hypothesis of TC development, normal follicular cells that accumulate errors can give rise to differentiated cancers, which in turn can develop into undifferentiated cancers following the enrichment of CSCs through the EMT process [13]. This is likely the reason why patients with ATCs, which consist of CSCs and non-CSCs, usually have a relapse after surgery and conventional chemotherapy and radio-iodine [15].
More recently, it has become clear that EMT is also involved in metabolic rewiring needed for the increased energetic demand of the mesenchymal cells compared to their epithelial counterparts due to the increased motility and invasion ability. In fact, EMT induction in epithelial mammary cells by Twist expression upregulates the expression of 44 metabolic genes, including dihydropyrimidine dehydrogenase (DPYD), an enzyme involved in pyrimidine catabolism, that in turn upregulates EMT [34]. Therefore, it is likely that metabolic rewiring is required for completeness of EMT. Other metabolic pathways modulated by EMT include glycolysis, lipid metabolism, mitochondrial metabolism and glutaminolysis. Specifically, it has been shown that EMT induction suppresses the expression of multiple metabolic proteins, including fructose-1.6-bisphosphatase 1 (FBP1), thus resulting in increased glycolysis [35], fatty acid synthase (FASN) and ACC, thus resulting in decreased lipogenesis [36], nucleoside transporter [37], and pyruvate dehydrogenase kinase 4 (PDK4) [38], whilst enhancing the expression of glutaminase 1 (GLS1) [39], enzymes of glutathione metabolism, cytochrome P450, aldehyde dehydrogenase, thus accounting for the increased chemoresistance [40], and GLUT3 [41]. On the other side, these metabolic alterations sustain the Warburg effect and induce EMT by enhancing glycolysis and blocking the TCA cycle. In particular, upregulation of (i) GLUT1 and GLUT3 glucose transporters activates matrix metallopeptidase MMP-2, which in turn induces EMT and invasiveness; (ii) HK1 and HK2 hexokinase, involved in the first step of glycolysis, activates Snail and Slug, which in turn induces EMT; (iii) PFKM and PFKP, rate-limiting enzymes of glycolysis, directly induce EMT; (iv) LDHA and LDHB, associated with enhanced glycolysis and lactate production, as well as extracellular lactate, activate Snail and therefore EMT [42].
References
- Hyeyeun Lim; Susan S. Devesa; Julie A. Sosa; David Check; Cari M. Kitahara; Trends in Thyroid Cancer Incidence and Mortality in the United States, 1974-2013. JAMA 2017, 317, 1338-1348, 10.1001/jama.2017.2719.
- Lola Rahib; Benjamin D. Smith; Rhonda Aizenberg; Allison B. Rosenzweig; Julie M. Fleshman; Lynn M. Matrisian; Projecting Cancer Incidence and Deaths to 2030: The Unexpected Burden of Thyroid, Liver, and Pancreas Cancers in the United States. Cancer Research 2014, 74, 2913-2921, 10.1158/0008-5472.can-14-0155.
- Wen‐Qi Bao; Hao Zi; Qian‐Qian Yuan; Lu‐Yao Li; Tong Deng; Global burden of thyroid cancer and its attributable risk factors in 204 countries and territories from 1990 to 2019. Thoracic Cancer 2021, 12, 2494-2503, 10.1111/1759-7714.14099.
- L. Dal Maso; M. Lise; P. Zambon; F. Falcini; E. Crocetti; Diego Serraino; C. Cirilli; R. Zanetti; Marina Vercelli; S. Ferretti; et al.F. StracciV. De LisiS. BuscoG. TagliabueM. BudroniR. TuminoA. GiacominS. Franceschi Incidence of thyroid cancer in Italy, 1991–2005: time trends and age–period–cohort effects. Annals of Oncology 2010, 22, 957-963, 10.1093/annonc/mdq467.
- Kimberly D. Miller Mph; Leticia Nogueira; Angela B. Mariotto; Julia H. Rowland; K. Robin Yabroff; Catherine M. Alfano; Ahmedin Jemal; Joan L. Kramer; Rebecca L. Siegel; Cancer treatment and survivorship statistics, 2019. CA: A Cancer Journal for Clinicians 2019, 69, 363-385, 10.3322/caac.21565.
- Somayyeh Fahiminiya; Leanne de Kock; William D Foulkes; Biologic and Clinical Perspectives on Thyroid Cancer. New England Journal of Medicine 2016, 375, 2306-2307, 10.1056/nejmc1613118.
- Furio Pacini; Which patient with thyroid cancer deserves systemic therapy and when?. Best Practice & Research Clinical Endocrinology & Metabolism 2017, 31, 291-294, 10.1016/j.beem.2017.08.001.
- Mohamed Aashiq; Deborah A. Silverman; Shorook Na’Ara; Hideaki Takahashi; Moran Amit; Radioiodine-Refractory Thyroid Cancer: Molecular Basis of Redifferentiation Therapies, Management, and Novel Therapies. Cancers 2019, 11, 1382, 10.3390/cancers11091382.
- Naiyarat Prasongsook; Aditi Kumar; Ashish Chintakuntlawar; Robert L Foote; Jan Kasperbauer; Julian Molina; Yolanda Garces; Daniel Ma; Michelle A Neben Wittich; Joseph Rubin; et al.Ronald RichardsonJohn MorrisIan HayVahab FatourechiBryan McIverMabel RyderGeoffrey ThompsonClive GrantMelanie RichardsThomas J SeboMichael RiveraVera SumanSarah M JenkinsRobert C SmallridgeKeith C Bible Survival in Response to Multimodal Therapy in Anaplastic Thyroid Cancer. The Journal of Clinical Endocrinology & Metabolism 2017, 102, 4506-4514, 10.1210/jc.2017-01180.
- Josip Ljubas; Therese Ovesen; Maria Rusan; A Systematic Review of Phase II Targeted Therapy Clinical Trials in Anaplastic Thyroid Cancer.. Cancers 2019, 11, 943, 10.3390/cancers11070943.
- Anastasios Maniakas; Ramona Dadu; Naifa L. Busaidy; Jennifer R. Wang; Renata Ferrarotto; Charles Lu; Michelle D. Williams; G. Brandon Gunn; Marie-Claude Hofmann; Gilbert Cote; et al.Jared SperlingNeil D. GrossErich M. SturgisRyan P. GoepfertStephen Y. LaiMaria E. CabanillasMark Zafereo Evaluation of Overall Survival in Patients With Anaplastic Thyroid Carcinoma, 2000-2019. JAMA Oncology 2020, 6, 1397, 10.1001/jamaoncol.2020.3362.
- Keith C. Bible; Electron Kebebew; James Brierley; Juan P. Brito; Maria E. Cabanillas; Thomas J. ClarkJr.; Antonio Di Cristofano; Robert Foote; Thomas Giordano; Jan Kasperbauer; et al.Kate NewboldYuri E. NikiforovGregory RandolphM. Sara RosenthalAnna M. SawkaManisha ShahAshok ShahaRobert SmallridgeCarol K. Wong-Clark 2021 American Thyroid Association Guidelines for Management of Patients with Anaplastic Thyroid Cancer. Thyroid 2021, 31, 337-386, 10.1089/thy.2020.0944.
- Terry F. Davies; Rauf Latif; Noga C. Minsky; Risheng Ma; The Emerging Cell Biology of Thyroid Stem Cells. The Journal of Clinical Endocrinology & Metabolism 2011, 96, 2692-2702, 10.1210/jc.2011-1047.
- A Borah; Sreejith Raveendran; A Rochani; Taira Maekawa; Sakthi Kumar; Targeting self-renewal pathways in cancer stem cells: clinical implications for cancer therapy. Oncogenesis 2015, 4, e177-e177, 10.1038/oncsis.2015.35.
- Z. Guo; H. Hardin; R. V. Lloyd; Cancer stem-like cells and thyroid cancer. Endocrine-Related Cancer 2014, 21, T285-T300, 10.1530/erc-14-0002.
- Joseph M. Curry; Madalina Tuluc; Diana Whitaker-Menezes; Julie A. Ames; Archana Anantharaman; Aileen Butera; Benjamin Leiby; David Cognetti; Federica Sotgia; Michael P. Lisanti; et al.Ubaldo E. Martinez-Outschoorn Cancer metabolism, stemness and tumor recurrence. Cell Cycle 2013, 12, 1371-1384, 10.4161/cc.24092.
- Sophia Y. Lunt; Matthew G. Vander Heiden; Aerobic Glycolysis: Meeting the Metabolic Requirements of Cell Proliferation. Annual Review of Cell and Developmental Biology 2011, 27, 441-464, 10.1146/annurev-cellbio-092910-154237.
- Matthew G. Vander Heiden; Lewis C. Cantley; Craig B. Thompson; Understanding the Warburg Effect: The Metabolic Requirements of Cell Proliferation. Science 2009, 324, 1029-1033, 10.1126/science.1160809.
- Rafael Moreno-Sánchez; Sara Rodriguez-Enriquez; Álvaro Marín-Hernández; Emma Saavedra; Energy metabolism in tumor cells. FEBS Journal 2007, 274, 1393-1418, 10.1111/j.1742-4658.2007.05686.x.
- Valeria R. Fantin; Julie St-Pierre; Philip Leder; Attenuation of LDH-A expression uncovers a link between glycolysis, mitochondrial physiology, and tumor maintenance. Cancer Cell 2006, 9, 425-434, 10.1016/j.ccr.2006.04.023.
- Ubaldo Martinez-Outschoorn; Federica Sotgia; Michael Lisanti; Power Surge: Supporting Cells “Fuel” Cancer Cell Mitochondria. Cell Metabolism 2012, 15, 4-5, 10.1016/j.cmet.2011.12.011.
- Stephanos Pavlides; Iset Vera; Ricardo Gandara; Sharon Sneddon; Richard G. Pestell; Isabelle Mercier; Ubaldo Martinez-Outschoorn; Diana Whitaker-Menezes; Anthony Howell; Federica Sotgia; et al.Michael P. Lisanti Warburg Meets Autophagy: Cancer-Associated Fibroblasts Accelerate Tumor Growth and Metastasis via Oxidative Stress, Mitophagy, and Aerobic Glycolysis. Antioxidants & Redox Signaling 2012, 16, 1264-1284, 10.1089/ars.2011.4243.
- Stephanos Pavlides; Diana Whitaker-Menezes; Remedios Castello-Cros; Neal Flomenberg; Agnieszka K. Witkiewicz; Philippe G. Frank; Mathew C. Casimiro; Chenguang Wang; Paolo Fortina; Sankar Addya; et al.Richard G. PestellUbaldo E. Martinez-OutschoornFederica SotgiaMichael P. Lisanti The reverse Warburg effect: Aerobic glycolysis in cancer associated fibroblasts and the tumor stroma. Cell Cycle 2009, 8, 3984-4001, 10.4161/cc.8.23.10238.
- Agnieszka K. Witkiewicz; Diana Whitaker-Menezes; Abhijit Dasgupta; Nancy J. Philp; Zhao Lin; Ricardo Gandara; Sharon Sneddon; Ubaldo E. Martinez-Outschoorn; Federica Sotgia; Michael P. Lisanti; et al. Using the “reverse Warburg effect” to identify high-risk breast cancer patients. Cell Cycle 2012, 11, 1108-1117, 10.4161/cc.11.6.19530.
- Ubaldo E. Martinez-Outschoorn; Zhao Lin; Casey Trimmer; Neal Flomenberg; Chenguang Wang; Stephanos Pavlides; Richard G. Pestell; Anthony Howell; Federica Sotgia; Michael P Lisanti; et al. Cancer cells metabolically “fertilize” the tumor microenvironment with hydrogen peroxide, driving the Warburg effect. Cell Cycle 2011, 10, 2504-2520, 10.4161/cc.10.15.16585.
- Pierre Sonveaux; Frédérique Végran; Thies Schroeder; Melanie C. Wergin; Julien Verrax; Zahid N. Rabbani; Christophe J. De Saedeleer; Kelly M. Kennedy; Caroline Diepart; Bénédicte F. Jordan; et al.Michael J. KelleyBernard GallezMiriam L. WahlOlivier FeronMark W. Dewhirst Targeting lactate-fueled respiration selectively kills hypoxic tumor cells in mice. Journal of Clinical Investigation 2008, 118, 3930-3942, 10.1172/jci36843.
- Ming Luo; Michael Brooks; Max S. Wicha; Asparagine and Glutamine: Co-conspirators Fueling Metastasis. Cell Metabolism 2018, 27, 947-949, 10.1016/j.cmet.2018.04.012.
- Raziyeh Abooshahab; Kourosh Hooshmand; S. Adeleh Razavi; Morteza Gholami; Maryam Sanoie; Mehdi Hedayati; Plasma Metabolic Profiling of Human Thyroid Nodules by Gas Chromatography-Mass Spectrometry (GC-MS)-Based Untargeted Metabolomics. Frontiers in Cell and Developmental Biology 2020, 8, 385, 10.3389/fcell.2020.00385.
- Yanan Xu; Xiaojiao Zheng; Yunping Qiu; Wei Jia; Jiadong Wang; Shankai Yin; Distinct Metabolomic Profiles of Papillary Thyroid Carcinoma and Benign Thyroid Adenoma. Journal of Proteome Research 2015, 14, 3315-3321, 10.1021/acs.jproteome.5b00351.
- Joseph A. Combs; Gina M. DeNicola; The Non-Essential Amino Acid Cysteine Becomes Essential for Tumor Proliferation and Survival. Cancers 2019, 11, 678, 10.3390/cancers11050678.
- Darya Buehler; Heather Hardin; Weihua Shan; Celina Montemayor-Garcia; Patrick S Rush; Sofia Asioli; Herbert Chen; Ricardo V Lloyd; Expression of epithelial-mesenchymal transition regulators SNAI2 and TWIST1 in thyroid carcinomas. Modern Pathology 2012, 26, 54-61, 10.1038/modpathol.2012.137.
- Robert G. Hardy; Carolina Vicente-Dueñas; Ines González-Herrero; Catriona Anderson; Teresa Flores; Sharon Hughes; Chris Tselepis; James A. Ross; Isidro Sanchez-Garcia; Snail Family Transcription Factors Are Implicated in Thyroid Carcinogenesis. The American Journal of Pathology 2007, 171, 1037-1046, 10.2353/ajpath.2007.061211.
- V. Vasko; A. V. Espinosa; W. Scouten; H. He; H. Auer; S. Liyanarachchi; Oleksandr Larin; V. Savchenko; G. L. Francis; A. de la Chapelle; et al.M. SajiM. D. Ringel Gene expression and functional evidence of epithelial-to-mesenchymal transition in papillary thyroid carcinoma invasion. Proceedings of the National Academy of Sciences 2007, 104, 2803-2808, 10.1073/pnas.0610733104.
- Yoav D. Shaul; Elizaveta Freinkman; William C. Comb; Jason R. Cantor; Wai Leong Tam; Prathapan Thiru; Dohoon Kim; Naama Kanarek; Michael E. Pacold; Walter W. Chen; et al.Brian BierieRichard PossematoFerenc ReinhardtRobert A. WeinbergMichael B. YaffeDavid M. Sabatini Dihydropyrimidine Accumulation Is Required for the Epithelial-Mesenchymal Transition. Cell 2014, 158, 1094-1109, 10.1016/j.cell.2014.07.032.
- Chenfang Dong; Tingting Yuan; Yadi Wu; Yifan Wang; Teresa W.M. Fan; Sumitra Miriyala; Yiwei Lin; Jun Yao; Jian Shi; Tiebang Kang; et al.Pawel LorkiewiczDaret St ClairMien-Chie HungB. Mark EversBinhua P. Zhou Loss of FBP1 by Snail-Mediated Repression Provides Metabolic Advantages in Basal-like Breast Cancer. Cancer Cell 2013, 23, 316-331, 10.1016/j.ccr.2013.01.022.
- Lei Jiang; Ling Xiao; Hidekazu Sugiura; Xiongfei Huang; Ahmed Atef Ahmed Ali; Makoto Kuro-O; Ralph J. DeBerardinis; David A. Boothman; Metabolic reprogramming during TGFβ1-induced epithelial-to-mesenchymal transition. Oncogene 2014, 34, 3908-3916, 10.1038/onc.2014.321.
- Xiaofeng Zheng; Julienne Carstens; Jiha Kim; Matthew Scheible; Judith Kaye; Hikaru Sugimoto; Chia-Chin Wu; Valerie S. LeBleu; Raghu Kalluri; Epithelial-to-mesenchymal transition is dispensable for metastasis but induces chemoresistance in pancreatic cancer. Nature 2015, 527, 525-530, 10.1038/nature16064.
- Yuting Sun; Anneleen Daemen; Georgia Hatzivassiliou; David Arnott; Catherine Wilson; Guanglei Zhuang; Min Gao; Peter Liu; Aaron Boudreau; Leisa Johnson; et al.Jeff Settleman Metabolic and transcriptional profiling reveals pyruvate dehydrogenase kinase 4 as a mediator of epithelial-mesenchymal transition and drug resistance in tumor cells. Cancer & Metabolism 2014, 2, 1-14, 10.1186/2049-3002-2-20.
- Danielle B. Ulanet; Kiley Couto; Abhishek Jha; Sung Choe; Amanda Wang; Hin-Koon Woo; Mya Steadman; Byron DelaBarre; Stefan Gross; Edward Driggers; et al.Marion DorschJonathan B. Hurov Mesenchymal Phenotype Predisposes Lung Cancer Cells to Impaired Proliferation and Redox Stress in Response to Glutaminase Inhibition. PLOS ONE 2014, 9, e115144, 10.1371/journal.pone.0115144.
- Kari R. Fischer; Anna Durrans; Sharrell Lee; Jianting Sheng; Fuhai Li; Stephen T. C. Wong; Hyejin Choi; Tyler El Rayes; Seongho Ryu; Juliane S Troeger; et al.Robert F. SchwabeLinda T. VahdatNasser K. AltorkiVivek MittalDingcheng Gao Epithelial-to-mesenchymal transition is not required for lung metastasis but contributes to chemoresistance. Nature 2015, 527, 472-476, 10.1038/nature15748.
- Mark Masin; Jessica Vazquez; Simona Rossi; Svenja Groeneveld; Natasha Samson; Petra C Schwalie; Bart Deplancke; Laura E Frawley; Jérôme Gouttenoire; Darius Moradpour; et al.Trudy G OliverEtienne Meylan GLUT3 is induced during epithelial-mesenchymal transition and promotes tumor cell proliferation in non-small cell lung cancer. Cancer & Metabolism 2014, 2, 11-11, 10.1186/2049-3002-2-11.
- Hyunkoo Kang; Hyunwoo Kim; Sungmin Lee; Hyesook Youn; Buhyun Youn; Role of Metabolic Reprogramming in Epithelial–Mesenchymal Transition (EMT). International Journal of Molecular Sciences 2019, 20, 2042, 10.3390/ijms20082042.


