 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Dennis Timmerman | + 2296 word(s) | 2296 | 2021-11-03 07:10:01 | | | |
| 2 | Amina Yu | Meta information modification | 2296 | 2021-11-04 03:39:02 | | | | |
| 3 | Amina Yu | Meta information modification | 2296 | 2021-11-05 07:01:44 | | |
Video Upload Options
Germ cell tumors (GCTs) are the most common solid malignancies in young men. Despite the high frequency of these cancers within this defined age group, the discovery of the exceptional sensitivity of these tumors to the platinum DNA crosslinking compound cisplatin has led to the survival of most patients, with the current five-year survival rate exceeding 95%.
1. Introduction
As GCTs are derived from embryonic germ cells, closely resembling embryonic stem cells, their hypersensitivity to DNA-damaging agents is often traced back to their early embryonic phenotype [1][2][3]; for instance, similarly to embryonic stem cells, GCTs often display a low/inefficient DNA damage response and, as opposed to most solid malignancies, GCTs that are naïve to systemic treatment rarely harbor TP53 mutations, irrespective of histology [4][5]. Moreover, the wild-type TP53 status of GCTs, combined with a pluripotent phenotype, high levels of PUMA and NOXA, and, often, low expression levels of CDKN1A (P21), result in a cellular disbalance and a favor towards apoptosis over DNA repair [6][7][8][9][10]. Furthermore, a physiological antagonist of P53, mouse double minute 2 homologue (MDM2), has been illustrated to be especially important in P53 regulation in GCTs, as it has been shown to hamper the apoptotic response via binding to P53 and can be a putative important clinical target [3][11][12][13][14]. It has already been shown that the inhibition of MDM2 and disruption of the MDM2–P53 interaction can potentiate apoptosis and sensitize GCT cells to cisplatin [11][12]. On the other hand, no correlation has been identified between the levels of MDM2 and the treatment response [5]. Furthermore, the existence of many MDM2 binding partners, and the reported synergy between MDM2 antagonists and (targeted) therapy, both in GCTs and other cancers, make this an interesting and relevant target as well [11][12][15][16]. Histologically and clinically, GCTs can be divided into two main subtypes, referring partly to their pluripotent potential, namely, seminomas and non-seminomas [1][2]. While patients presenting with seminomas have an excellent prognosis, patients harboring non-seminomas have a mixed prognosis, based on tumor histology (e.g., embryonal carcinoma (EC), yolk sac tumor (YST), choriocarcinoma (CC), or teratoma (TE)), therapy naivety or chemotherapeutic resistance, and anatomical location, mainly focusing on extra-cranial GCTs of the mediastinum versus the testis [1][2][4][9][17]. Apart from tumor histology and origin, the P53 pathway and deregulation thereof has been studied in light of GCT treatment resistance [3][4][5][8][9][11][12][14][18]. Even though P53′s have many implications in resistance, no clear-cut result has been obtained that displays their role in clinical resistance, especially related to informative in vitro models [5][18]. In this entry, we focused on the latter (i.e., mediastinal GCTs vs. testicular GCTs) and developed a novel approach to shed light on the difference in treatment resistance between testicular and mediastinal GCTs. This is an important issue, as it is currently unclear whether mediastinal GCTs are more resistant to treatment because of their TP53 mutations, or whether these mutations simply occur more in these tumors as these tumors harbor different intrinsic resistance mechanisms.
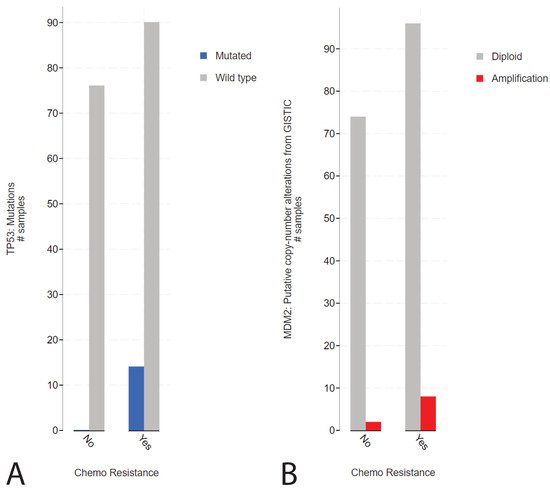
2. Presence of TP53 Mutations in Refractory Cisplatin-Resistant GCTs with a Specificity towards Mediastinal Localization
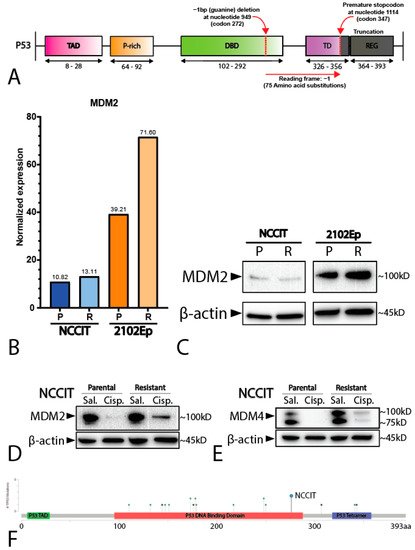
3. Mediastinal GCT Cell Line NCCIT Harbors Low Levels of MDM2 and Mutant TP53 whereas Testicular GCT Cell Line 2102Ep Harbors Wild-Type TP53 and High Levels of MDM2
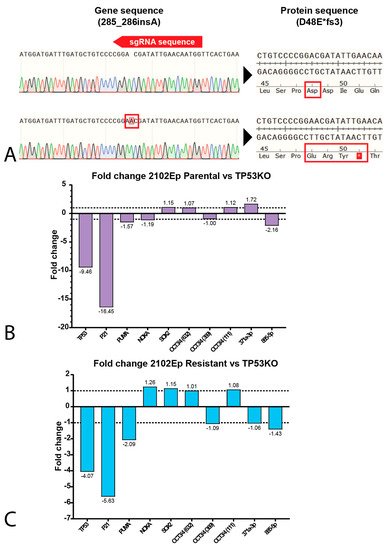
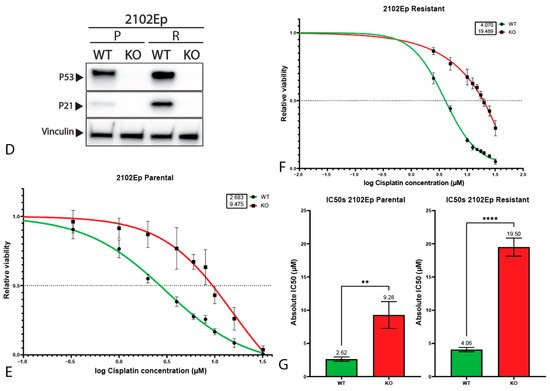
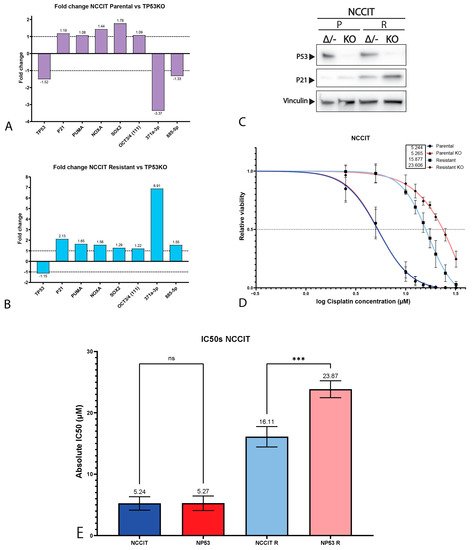
4. P53 Is Involved in Cisplatin Resistance in Both Wild-Type (Testicular) and Mutant (Mediastinal) GCT Cell Lines
References
- Oosterhuis, J.W.; Looijenga, L. Testicular Germ-Cell Tumours in a Broader Perspective. Nat. Rev. Cancer 2005, 5, 210–222.
- Oosterhuis, J.W.; Looijenga, L.H.J. Human Germ Cell Tumours from a Developmental Perspective. Nat. Rev. Cancer 2019, 19, 522–537.
- Timmerman, D.; Remmers, T.; Hillenius, S.; Looijenga, L. Mechanisms of TP53 Pathway Inactivation in Embryonic and Somatic Cells—Relevance for Understanding (Germ Cell) Tumorigenesis. Int. J. Mol. Sci. 2021, 22, 5377.
- Bagrodia, A.; Lee, B.; Lee, W.; Cha, E.K.; Sfakianos, J.P.; Iyer, G.; Pietzak, E.J.; Gao, S.P.; Zabor, E.C.; Ostrovnaya, I.; et al. Genetic Determinants of Cisplatin Resistance in Patients with Advanced Germ Cell Tumors. J. Clin. Oncol. 2016, 34, 4000–4007.
- Kersemaekers, A.-M.F.; Mayer, F.; Molier, M.; Van Weeren, P.C.; Oosterhuis, J.W.; Bokemeyer, C.; Looijenga, L.H. Role of P53 and MDM2 in Treatment Response of Human Germ Cell Tumors. J. Clin. Oncol. 2002, 20, 1551–1561.
- Ulbright, T.M.; Orazi, A.; De Riese, W.; De Riese, C.; Messemer, E.J.; Foster, R.S.; Donohue, J.P.; Eble, J.N. The Correlation of P53 Protein Expression with Proliferative Activity and Occult Metastases in Clinical Stage I Non-Seminomatous Germ Cell Tumors of the Testis. Mod. Pathol. 1994, 7, 64–68.
- Filion, T.M.; Qiao, M.; Ghule, P.N.; Mandeville, M.; van Wijnen, A.J.; Stein, J.L.; Lian, J.B.; Altieri, D.C.; Stein, G.S. Survival Responses of Human Embryonic Stem Cells to DNA Damage. J. Cell. Physiol. 2009, 220, 586–592.
- Gutekunst, M.; Oren, M.; Weilbacher, A.; Dengler, M.A.; Markwardt, C.; Thomale, J.; Aulitzky, W.E.; Van Der Kuip, H. p53 Hypersensitivity is the Predominant Mechanism of the Unique Responsiveness of Testicular Germ Cell Tumor (TGCT) Cells to Cisplatin. PLoS ONE 2011, 6, e19198.
- Jacobsen, C.; Honecker, F. Cisplatin Resistance in Germ Cell Tumours: Models and Mechanisms. Andrology 2015, 3, 111–121.
- Bloom, J.C.; Loehr, A.R.; Schimenti, J.C.; Weiss, R.S. Germline Genome Protection: Implications for Gamete Quality and Germ Cell Tumorigenesis. Andrology 2019, 7, 516–526.
- Bauer, S.; Mühlenberg, T.; Leahy, M.; Hoiczyk, M.; Gauler, T.; Schuler, M.; Looijenga, L. Therapeutic Potential of Mdm2 Inhibition in Malignant Germ Cell Tumours. Eur. Urol. 2010, 57, 679–687.
- Koster, R.; Timmer-Bosscha, H.; Bischoff, R.; Gietema, J.; De Jong, S. Disruption of the MDM2–p53 Interaction Strongly Potentiates p53-Dependent Apoptosis in Cisplatin-Resistant Human Testicular Carcinoma Cells via the Fas/FasL Pathway. Cell Death Dis. 2011, 2, e148.
- Gutekunst, M.; Mueller, T.; Weilbacher, A.; Dengler, M.A.; Bedke, J.; Kruck, S.; Oren, M.; Aulitzky, W.E.; Van Der Kuip, H. Cisplatin Hypersensitivity of Testicular Germ Cell Tumors Is Determined by High Constitutive Noxa Levels Mediated by Oct-4. Cancer Res. 2013, 73, 1460–1469.
- Lobo, J.; Jerónimo, C.; Henrique, R. Cisplatin Resistance in Testicular Germ Cell Tumors: Current Challenges from Various Perspectives. Cancers 2020, 12, 1601.
- Mego, M.; Van Agthoven, T.; Gronesova, P.; Chovanec, M.; Miskovska, V.; Mardiak, J.; Looijenga, L.H.J. Clinical Utility of Plasma miR-371a-3p in Germ Cell Tumors. J. Cell. Mol. Med. 2019, 23, 1128–1136.
- Fåhraeus, R.; Olivares-Illana, V. MDM2’s Social Network. Oncogene 2013, 33, 4365–4376.
- Zhou, Z.-T.; Wang, J.-W.; Yang, L.; Wang, J.; Zhang, W. Primary Germ Cell Tumor in the Mediastinum-Report of 47 Cases. Zhonghua Zhong Liu Za Zhi 2006, 28, 863–866.
- Burger, H.; Nooter, K.; Boersma, A.W.; Kortland, C.J.; Stoter, G. Lack of Correlation Between Cisplatin-Induced Apoptosis, p53 Status and Expression of Bcl-2 Family Proteins in Testicular Germ Cell Tumour Cell Lines. Int. J. Cancer 1997, 73, 592–599.
- Cerami, E.; Gao, J.; Dogrusoz, U.; Gross, B.E.; Sumer, S.O.; Aksoy, B.A.; Jacobsen, A.; Byrne, C.J.; Heuer, M.L.; Larsson, E.; et al. The cBio Cancer Genomics Portal: An Open Platform for Exploring Multidimensional Cancer Genomics Data: Figure 1. Cancer Discov. 2012, 2, 401–404.
- Gao, J.; Aksoy, B.A.; Dogrusoz, U.; Dresdner, G.; Gross, B.; Sumer, S.O.; Sun, Y.; Jacobsen, A.; Sinha, R.; Larsson, E.; et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci. Signal. 2013, 6, l1.
- de Jong, J.; Stoop, H.; Gillis, A.J.M.; Hersmus, R.; van Gurp, R.J.H.L.M.; van de Geijn, G.-J.M.; van Drunen, E.; Beverloo, H.B.; Schneider, D.T.; Sherlock, J.K.; et al. Further Characterization of the First Seminoma Cell Line TCam-2. Genes Chromosom. Cancer 2008, 47, 185–196.
- Li, J.; Kurokawa, M. Regulation of MDM2 Stability After DNA Damage. J. Cell. Physiol. 2015, 230, 2318–2327.
- Spierings, D.; De Vries, E.E.G.; Vellenga, E.; De Jong, S. The Attractive Achilles Heel of Germ Cell Tumours: An Inherent Sensitivity to Apoptosis-Inducing Stimuli. J. Pathol. 2003, 200, 137–148.
- Kerley-Hamilton, J.S.; Pike, A.M.; Li, N.; DiRenzo, J.; Spinella, M.J. A p53-Dominant Transcriptional Response to Cisplatin in Testicular Germ Cell Tumor-Derived Human Embyronal Carcinoma. Oncogene 2005, 24, 6090–6100.
- Romano, F.J.; Rossetti, S.; Conteduca, V.; Schepisi, G.; Cavaliere, C.; Di Franco, R.; La Mantia, E.; Castaldo, L.; Nocerino, F.; Ametrano, G.; et al. Role of DNA Repair Machinery and P53 in the Testicular Germ Cell Cancer: A Review. Oncotarget 2016, 7, 85641–85649.
- Baugh, E.H.; Ke, H.; Levine, A.J.; Bonneau, A.R.; Chan, C.S. Why are there Hotspot Mutations in the TP53 Gene in Human Cancers? Cell Death Differ. 2018, 25, 154–160.
- Voorhoeve, P.M.; le Sage, C.; Schrier, M.; Gillis, A.J.; Stoop, H.; Nagel, R.; Liu, Y.-P.; van Duijse, J.; Drost, J.; Griekspoor, A.; et al. A Genetic Screen Implicates miRNA-372 and miRNA-373 as Oncogenes in Testicular Germ Cell Tumors. Cell 2006, 124, 1169–1181.
- Lobo, J.; Gillis, A.J.; Berg, A.V.D.; Dorssers, L.C.J.; Belge, G.; Dieckmann, K.-P.; Roest, H.P.; Van Der Laan, L.J.W.; Gietema, J.; Hamilton, R.J.; et al. Identification and Validation Model for Informative Liquid Biopsy-Based microRNA Biomarkers: Insights from Germ Cell Tumor In Vitro, In Vivo and Patient-Derived Data. Cells 2019, 8, 1637.
- Almstrup, K.; Lobo, J.; Mørup, N.; Belge, G.; Meyts, E.R.-D.; Looijenga, L.H.J.; Dieckmann, K.-P. Application of miRNAs in the Diagnosis and Monitoring of Testicular Germ Cell Tumours. Nat. Rev. Urol. 2020, 17, 201–213.
- Murray, M.J.; Bell, E.; Raby, K.L.; Rijlaarsdam, M.; Gillis, A.J.M.; Looijenga, L.; Brown, H.; Destenaves, B.; Nicholson, J.C.; Coleman, N.S. A pipeline to Quantify Serum and Cerebrospinal Fluid Micrornas for Diagnosis and Detection of Relapse in Paediatric Malignant Germ-Cell Tumours. Br. J. Cancer 2016, 114, 151–162.
- Murray, M.J.; Halsall, D.J.; Hook, C.E.; Williams, D.M.; Nicholson, J.C.; Coleman, N.; Sweet, W.; Duh, Y.-J.; Greenfield, L.; Tarco, E.; et al. Identification of MicroRNAs from the miR-371∼373 and miR-302 Clusters as Potential Serum Biomarkers of Malignant Germ Cell Tumors. Am. J. Clin. Pathol. 2011, 135, 119–125.
- Syring, I.; Bartels, J.; Holdenrieder, S.; Kristiansen, G.; Müller, S.C.; Ellinger, J. Circulating Serum miRNA (miR-367-3p, miR-371a-3p, miR-372-3p and miR-373-3p) as Biomarkers in Patients with Testicular Germ Cell Cancer. J. Urol. 2015, 193, 331–337.
- Leão, R.; Albersen, M.; Looijenga, L.H.; Tandstad, T.; Kollmannsberger, C.; Murray, M.J.; Culine, S.; Coleman, N.; Belge, G.; Hamilton, R.J.; et al. Circulating MicroRNAs, the Next-Generation Serum Biomarkers in Testicular Germ Cell Tumours: A Systematic Review. Eur. Urol. 2021, 80, 456–466.


