Germ cell tumors (GCTs) are the most common solid malignancies in young men. Despite the high frequency of these cancers within this defined age group, the discovery of the exceptional sensitivity of these tumors to the platinum DNA crosslinking compound cisplatin has led to the survival of most patients, with the current five-year survival rate exceeding 95%.
- human malignant germ cell tumors
- mediastinal germ cell tumors
- testicular germ cell tumors
- TP53
1. Introduction
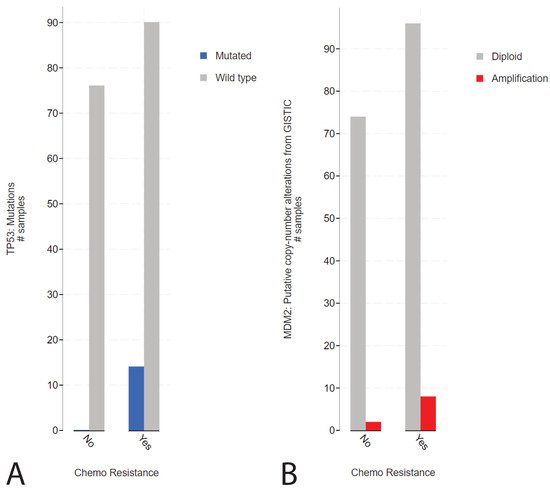
As GCTs are derived from embryonic germ cells, closely resembling embryonic stem cells, their hypersensitivity to DNA-damaging agents is often traced back to their early embryonic phenotype [1][2][3][6,7,8]; for instance, similarly to embryonic stem cells, GCTs often display a low/inefficient DNA damage response and, as opposed to most solid malignancies, GCTs that are naïve to systemic treatment rarely harbor TP53 mutations, irrespective of histology [4][5][9,10]. Moreover, the wild-type TP53 status of GCTs, combined with a pluripotent phenotype, high levels of PUMA and NOXA, and, often, low expression levels of CDKN1A (P21), result in a cellular disbalance and a favor towards apoptosis over DNA repair [6][7][8][9][10][11,12,13,14,15]. Furthermore, a physiological antagonist of P53, mouse double minute 2 homologue (MDM2), has been illustrated to be especially important in P53 regulation in GCTs, as it has been shown to hamper the apoptotic response via binding to P53 and can be a putative important clinical target [3][11][12][13][14][8,16,17,18,19]. It has already been shown that the inhibition of MDM2 and disruption of the MDM2–P53 interaction can potentiate apoptosis and sensitize GCT cells to cisplatin [11][12][16,17]. On the other hand, no correlation has been identified between the levels of MDM2 and the treatment response [5][10]. Furthermore, the existence of many MDM2 binding partners, and the reported synergy between MDM2 antagonists and (targeted) therapy, both in GCTs and other cancers, make this an interesting and relevant target as well [11][12][15][16][16,17,20,21]. Histologically and clinically, GCTs can be divided into two main subtypes, referring partly to their pluripotent potential, namely, seminomas and non-seminomas [1][2][6,7]. While patients presenting with seminomas have an excellent prognosis, patients harboring non-seminomas have a mixed prognosis, based on tumor histology (e.g., embryonal carcinoma (EC), yolk sac tumor (YST), choriocarcinoma (CC), or teratoma (TE)), therapy naivety or chemotherapeutic resistance, and anatomical location, mainly focusing on extra-cranial GCTs of the mediastinum versus the testis [1][2][4][9][17][6,7,9,14,22]. Apart from tumor histology and origin, the P53 pathway and deregulation thereof has been studied in light of GCT treatment resistance [3][4][5][8][9][11][12][14][18][8,9,10,13,14,16,17,19,23]. Even though P53′s have many implications in resistance, no clear-cut result has been obtained that displays their role in clinical resistance, especially related to informative in vitro models [5][18][10,23]. In this enstrudy, we focused on the latter (i.e., mediastinal GCTs vs. testicular GCTs) and developed a novel approach to shed light on the difference in treatment resistance between testicular and mediastinal GCTs. This is an important issue, as it is currently unclear whether mediastinal GCTs are more resistant to treatment because of their TP53 mutations, or whether these mutations simply occur more in these tumors as these tumors harbor different intrinsic resistance mechanisms.
2. Presence of TP53 Mutations in Refractory Cisplatin-Resistant GCTs with a Specificity towards Mediastinal Localization
1. Presence of TP53 Mutations in Refractory Cisplatin-Resistant GCTs with a Specificity towards Mediastinal Localization
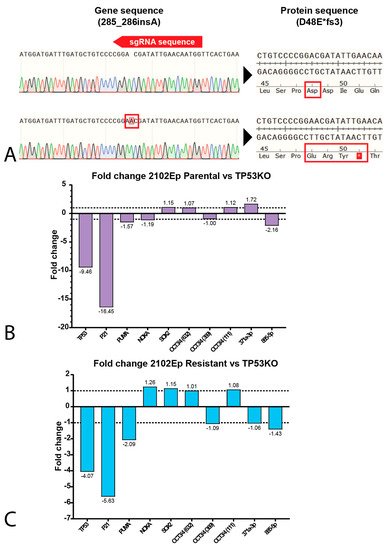
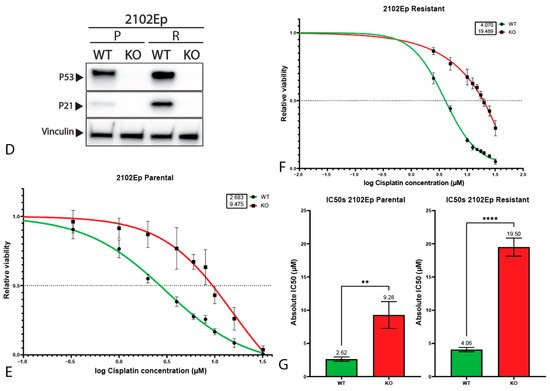
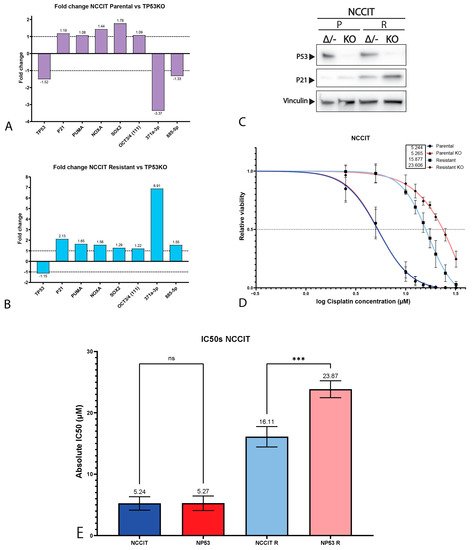
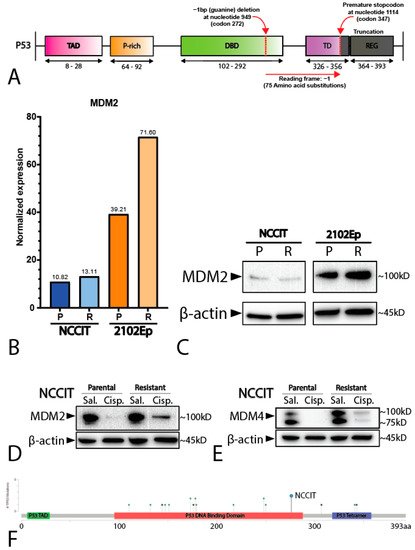
32. Mediastinal GCT Cell Line NCCIT Harbors Low Levels of MDM2 and Mutant TP53 whereas Testicular GCT Cell Line 2102Ep Harbors Wild-Type TP53 and High Levels of MDM2
43. P53 Is Involved in Cisplatin Resistance in Both Wild-Type (Testicular) and Mutant (Mediastinal) GCT Cell Lines