 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Margherita Sisto | + 4280 word(s) | 4280 | 2021-08-02 10:23:34 | | | |
| 2 | Vivi Li | Meta information modification | 4280 | 2021-08-03 11:24:02 | | |
Video Upload Options
For decades, metalloproteinase 17 (ADAM17) has been the goal of wide investigation. Since its discovery as the tumour necrosis factor-α convertase, it has been studied as the main drug target, especially in the context of inflammatory conditions and tumour. In fact, evidence is mounting to support a key role of ADAM17 in the induction of the proliferation, migration and progression of tumour cells and the trigger of the pro-fibrotic process during chronic inflammatory conditions; this occurs, probably, through the activation of epithelial-to-mesenchymal transition (EMT). EMT is a central morphologic conversion that occurs in adults during wound healing, tumour progression and organ fibrosis. EMT is characterised by the disassembly of cell–cell contacts, remodelling of the actin cytoskeleton and separation of cells, and generates fibroblast-like cells that express mesenchymal markers and have migratory properties. This transition is characterised by loss of epithelial proteins such as E-cadherin and the acquisition of new mesenchymal markers, including vimentin and a-smooth muscle actin.
1. Introduction
2. The Sheddase ADAM17: Biology and Function
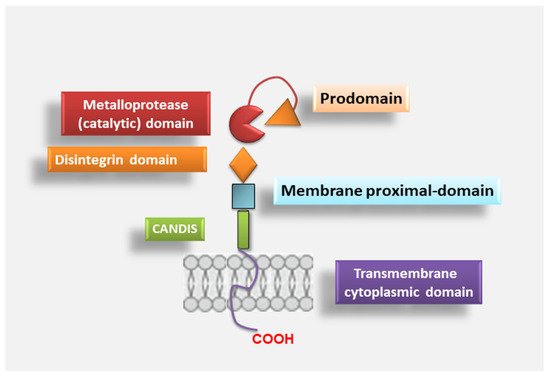
2.1. ADAM17 Structure
2.2. ADAM17 Activation
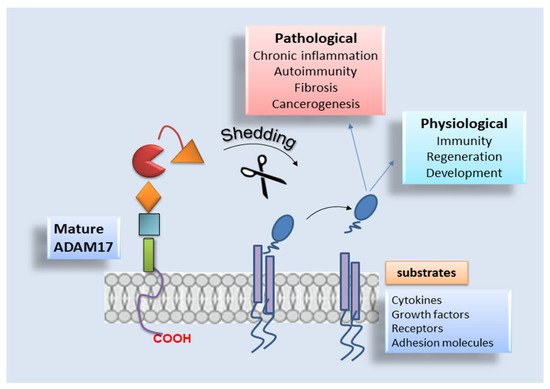
2.3. ADAM17 Distribution and Substrates
3. Mechanism of ADAM17 Signals Modulation in Fibrotic Diseases and Cancer
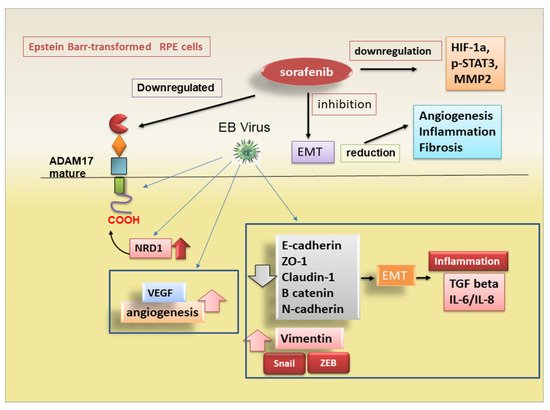
3.1. ADAM17-Mediated Regulation of EMT in Degenerative Retinopathy
3.2. Pro-Fibrotic Activity of ADAM17 in Diabetic Nephropaty
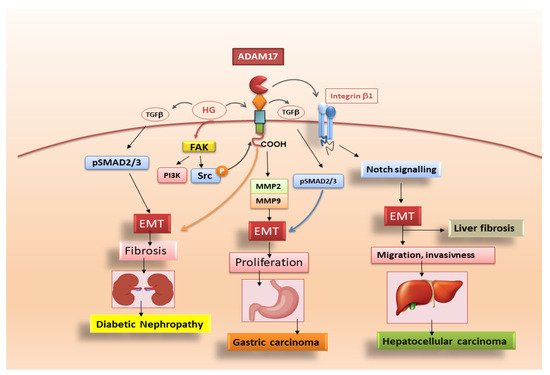
3.3. Adam17 Promotes EMT in Gastric Carcinoma
3.4. ADAM17-Mediated Mechanisms in Liver Fibrosis
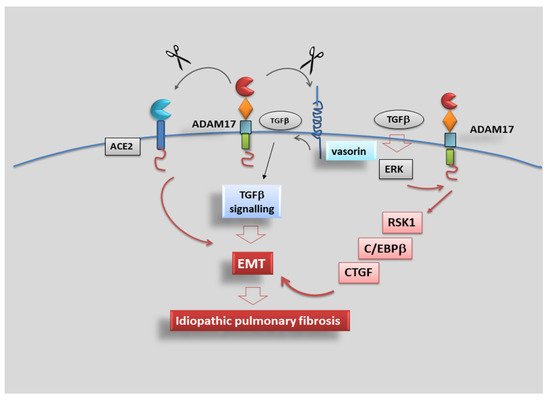
3.5. Role of ADAM17 in Idiopathic Pulmonary Fibrosis
References
- Lichtenthaler, S.F.; Lemberg, M.K.; Fluhrer, R. Proteolytic ectodomain shedding of membrane proteins in mammals-hardware, concepts, and recent developments. EMBO J. 2018, 37, e99456.
- Calligaris, M.; Cuffaro, D.; Bonelli, S.; Spanò, D.P.; Rossello, A.; Nuti, E.; Scilabra, S.D. Strategies to Target ADAM17 in Disease: From its Discovery to the iRhom Revolution. Molecules 2021, 26, 944.
- Grötzinger, J.; Lorenzen, I.; Düsterhöft, S. Molecular insights into the multilayered regulation of ADAM17: The role of the extracellular region. Biochim. Biophys. Acta Mol. Cell. Res. 2017, 1864, 2088–2095.
- Lambrecht, B.N.; Vanderkerken, M.; Hammad, H. The emerging role of ADAM metalloproteinases in immunity. Nat. Rev. Immunol. 2018, 18, 745–758.
- Schmidt-Arras, D.; Rose-John, S. Regulation of Fibrotic Processes in the Liver by ADAM Proteases. Cells 2019, 8, 1226.
- Lisi, S.; D’Amore, M.; Sisto, M. ADAM17 at the interface between inflammation and autoimmunity. Immunol. Lett. 2014, 162, 159–169.
- Xu, M.; Zhou, H.; Zhang, C.; He, J.; Wei, H.; Zhou, M.; Lu, Y.; Sun, Y.; Ding, J.W.; Zeng, J.; et al. ADAM17 promotes epithelial-mesenchymal transition via TGF-β/Smad pathway in gastric carcinoma cells. Int. J. Oncol. 2016, 49, 2520–2528.
- Malapeira, J.; Esselens, C.; Bech-Serra, J.J.; Canals, F.; Arribas, J. ADAM17 (TACE) regulates TGFβ signaling through the cleavage of vasorin. Oncogene 2011, 30, 1912–1922.
- Hsia, H.E.; Tüshaus, J.; Brummer, T.; Zheng, Y.; Scilabra, S.D.; Lichtenthaler, S.F. Functions of ‘A disintegrin and metalloproteases (ADAMs)’ in the mammalian nervous system. Cell. Mol. Life Sci. 2019, 76, 3055–3081.
- Blobel, C.P. ADAMs: Key components in EGFR signalling and development. Nat. Rev. Mol. Cell. Biol. 2005, 6, 32–43.
- Edwards, D.R.; Handsley, M.M.; Pennington, C.J. The ADAM metalloproteinases. Mol. Aspects Med. 2008, 29, 258–289.
- Zunke, F.; Rose-John, S. The shedding protease ADAM17: Physiology and pathophysiology. Biochim. Biophys. Acta Mol. Cell Res. 2017, 1864, 2059–2070.
- Brocker, C.N.; Vasiliou, V.; Nebert, D.W. Evolutionary divergence and functions of the ADAM and ADAMTS gene families. Hum. Genom. 2009, 4, 43–55.
- Black, R.A.; Rauch, C.T.; Kozlosky, C.J.; Peschon, J.J.; Slack, J.L.; Wolfson, M.F.; Castner, B.J.; Stocking, K.L.; Reddy, P.; Srinivasan, S.; et al. A metalloproteinase disintegrin that releases tumour-necrosis factor-alpha from cells. Nature 1997, 385, 729–733.
- Moss, M.L.; Jin, S.L.; Milla, M.E.; Bickett, D.M.; Burkhart, W.; Carter, H.L.; Chen, W.-J.; Clay, W.C.; Didsbury, J.R.; Hassler, D.; et al. Cloning of a disintegrin metalloproteinase that processes precursor tumour-necrosis factor-alpha. Nature 1997, 385, 733–736.
- Black, R.A.; White, J.M. ADAMs: Focus on the protease domain. Curr. Opin. Cell Biol. 1998, 10, 654–659.
- Blobel, C.P. Metalloprotease-disintegrins: Links to cell adhesion and cleavage of TNF alpha and Notch. Cell 1997, 90, 589–592.
- Lorenzen, I.; Lokau, J.; Korpys, Y.; Oldefest, M.; Flynn, C.M.; Künzel, U.; Garbers, C.; Freeman, M.; Grötzinger, J.; Düsterhöft, S. Control of ADAM17 activity by regulation of its cellular localisation. Sci. Rep. 2016, 6, 35067.
- Galazka, G.; Windsor, L.J.; Birkedal-Hansen, H.; Engler, J.A. APMA (4-aminophenylmercuric acetate) activation of stromelysin-1 involves protein interactions in addition to those with cysteine-75 in the propeptide. Biochemistry 1996, 35, 11221–11227.
- Van Wart, H.E.; Birkedal-Hansen, H. The cysteine switch: A principle of regulation of metalloproteinase activity with potential applicability to the entire matrix metalloproteinase gene family. Proc. Natl. Acad. Sci. USA 1990, 87, 5578–5582.
- Roghani, M.; Becherer, J.D.; Moss, M.L.; Atherton, R.E.; Erdjument-Bromage, H.; Arribas, J.; Blackburn, R.K.; Weskamp, G.; Tempst, P.; Blobel, C.P.; et al. Metalloprotease-disintegrin MDC9: Intracellular maturation and catalytic activity. J. Biol. Chem. 1999, 274, 3531–3540.
- Reiss, K.; Saftig, P. The “a disintegrin and metalloprotease” (ADAM) family of sheddases: Physiological and cellular functions. Semin. Cell Dev. Biol. 2009, 20, 126–137.
- Schlöndorff, J.; Becherer, J.D.; Blobel, C.P. Intracellular maturation and localization of the tumour necrosis factor alpha convertase (TACE). Biochem. J. 2000, 347, 131–138.
- Lisi, S.; Sisto, M.; Lofrumento, D.D.; Cucci, L.; Frassanito, M.A.; Mitolo, V.; D’Amore, M. Pro-inflammatory role of Anti-Ro/SSA autoantibodies through the activation of Furin-TACE-amphiregulin axis. J. Autoimmun. 2010, 35, 160–170.
- Adrain, C.; Freeman, M. New lives for old: Evolution of pseudoenzyme function illustrated by iRhoms. Nat. Rev. Mol. Cell Biol. 2012, 13, 489–498.
- Dreymueller, D.; Pruessmeyer, J.; Groth, E.; Ludwig, A. The role of ADAM-mediated shedding in vascular biology. Eur. J. Cell Biol. 2012, 91, 472–485.
- Tellier, E.; Canault, M.; Rebsomen, L.; Bonardo, B.; Juhan-Vague, I.; Nalbone, G.; Peiretti, F. The shedding activity of ADAM17 is sequestered in lipid rafts. Exp. Cell Res. 2006, 312, 3969–3980.
- Srour, N.; Lebel, A.; McMahon, S.; Fournier, I.; Fugère, M.; Day, R.; Dubois, C.M. TACE/ADAM-17 maturation and activation of sheddase activity require proprotein convertase activity. FEBS Lett. 2003, 554, 275–283.
- Zhang, P.; Shen, M.; Fernandez-Patron, C.; Kassiri, Z. ADAMs family and relatives in cardiovascular physiology and pathology. J. Mol. Cell. Cardiol. 2016, 93, 186–199.
- Sommer, A.; Kordowski, F.; Buch, J.; Maretzky, T.; Evers, A.; Andrä, J.; Düsterhöft, S.; Michalek, M.; Lorenzen, I.; Somasundaram, P.; et al. Phosphatidylserine exposure is required for ADAM17 sheddase function. Nat. Commun. 2016, 7, 11523.
- Dulloo, I.; Muliyil, S.; Freeman, M. The molecular, cellular and pathophysiological roles of iRhom pseudoproteases. Open Biol. 2019, 9, 190003.
- Düsterhöft, S.; Lokau, J.; Garbers, C. The metalloprotease ADAM17 in inflammation and cancer. Pathol. Res. Pract. 2019, 215, 152410.
- Lichtenthaler, B.F.; O’Hara, C.P. Blobel iRhoms in the brain—A new frontier? Cell Cycle 2015, 14, 3003–3004.
- Maretzky, T.; McIlwain, D.R.; Issuree, P.D.; Li, X.; Malapeira, J.; Amin, S.; Lang, P.A.; Mak, T.W.; Blobel, C.P. iRhom2 controls the substrate selectivity of stimulated ADAM17-dependent ectodomain shedding. Proc. Natl. Acad. Sci. USA 2013, 110, 433–438.
- Lisi, S.; Sisto, M.; Lofrumento, D.D.; D’Amore, M. Sjögren’s syndrome autoantibodies provoke changes in gene expression profiles of inflammatory cytokines triggering a pathway involving TACE/NF-κB. Lab. Investig. 2012, 92, 615–624.
- Sisto, M.; Lisi, S.; Lofrumento, D.D.; D’Amore, M.; Frassanito, M.A.; Ribatti, D. Sjögren’s syndrome pathological neovascularization is regulated by VEGF-A-stimulated TACE-dependent crosstalk between VEGFR2 and NF-κB. Genes Immun. 2012, 13, 411–420.
- Lee, A.M.; Diasio, R.B. ADAM-17: A target to increase chemotherapeutic efficacy in colorectal cancer? Clin. Cancer Res. 2010, 16, 3319–3321.
- Peschon, J.J.; Slack, J.L.; Reddy, P.; Stocking, K.L.; Sunnarborg, S.W.; Lee, D.C.; Russell, W.E.; Castner, B.J.; Johnson, R.S.; Fitzner, J.N.; et al. An essential role for ectodomain shedding in mammalian development. Science 1998, 282, 1281–1284.
- Jackson, L.F.; Qiu, T.H.; Sunnarborg, S.W.; Chang, A.; Zhang, C.; Patterson, C.; Lee, D.C. Defective valvulogenesis in HB-EGF and TACE-null mice is associated with aberrant BMP signaling. EMBO J. 2003, 22, 2704–2716.
- Bandsma, R.H.; van Goor, H.; Yourshaw, M.; Horlings, R.K.; Jonkman, M.F.; Schölvinck, E.H.; Karrenbeld, A.; Scheenstra, R.; Komhoff, M.; Rump, P.; et al. Loss of ADAM17 is associated with severe multiorgan dysfunction. Hum. Pathol. 2015, 46, 923–928.
- Shen, H.; Li, L.; Zhou, S.; Yang, S.; Chen, X.; Wang, D.; Zhong, S.; Zhao, J.; Tang, J. The role of ADAM17 in tumorigenesis and progression of breast cancer. Tumor Biol. 2016, 37, 15359–15370.
- Moss, M.L.; Minond, D. Recent Advances in ADAM17 Research: A Promising Target for Cancer and Inflammation. Mediators Inflamm. 2017, 2017, 9673537.
- Schmidt, S.; Schumacher, N.; Schwarz, J.; Tangermann, S.; Kenner, L.; Schlederer, M.; Sibilia, M.; Linder, M.; Altendorf-Hofmann, A.; Knösel, T.; et al. ADAM17 is required for EGF-R–induced intestinal tumors via IL-6 trans-signaling. J. Exp. Med. 2018, 215, 1205–1225.
- Dosch, J.; Ziemke, E.; Wan, S.; Luker, K.; Welling, T.; Hardiman, K.; Fearon, E.; Thomas, S.; Flynn, M.; Rios-Doria, J.; et al. Targeting ADAM17 inhibits human colorectal adenocarci-noma progression and tumor-initiating cell frequency. Oncotarget 2017, 8, 65090.
- Das, S.; Czarnek, M.; Bzowska, M.; Mezyk-Kope´c, R.; Stalinska, K.; Wyroba, B.; Sroka, J.; Jucha, J.; Deneka, D.; Stoklova, P.; et al. ADAM17 Silencing in Mouse Colon Carcino-ma Cells: The Effect on Tumoricidal Cytokines and Angiogenesis. PLoS ONE 2012, 7, e50791.
- Giricz, O.; Calvo, V.; Peterson, E.A.; Abouzeid, C.M.; Kenny, P.A. TACE-dependent TGFα shedding drives triple-negative breast cancer cell invasion. Int. J. Cancer 2013, 133, 2587–2595.
- Gao, M.Q.; Kim, B.G.; Kang, S.; Choi, Y.P.; Yoon, J.H.; Cho, N.H. Human breast cancer-associated fibroblasts enhance cancer cell pro-liferation through increased TGF-α cleavage by ADAM17. Cancer Lett. 2013, 336, 240–246.
- Chalaris, A.; Adam, N.; Sina, C.; Rosenstiel, P.; Lehmann-Koch, J.; Schirmacher, P.; Hartmann, D.; Cichy, J.; Gavrilova, O.; Schreiber, S.; et al. Critical role of the disintegrin metalloprotease ADAM17 for intestinal inflammation and regeneration in mice. J. Exp. Med. 2010, 207, 1617–1624.
- Park, G.B.; Kim, D.; Kim, Y.S.; Kim, J.W.; Sun, H.; Roh, K.H.; Yang, J.W.; Hur, D.Y. Regulation of ADAM10 and ADAM17 by Sorafenib Inhibits Epithelial-to-Mesenchymal Transition in Epstein-Barr Virus-Infected Retinal Pigment Epithelial Cells. Investig. Ophthalmol. Vis. Sci. 2015, 56, 5162–5173.
- Pastor, J.C.; de la Rua, E.R.; Martın, F. Proliferative vitreoretinopathy: Risk factors and pathobiology. Prog. Retin Eye Res. 2002, 21, 127–144.
- Tamiya, S.; Liu, L.; Kaplan, H.J. Epithelial-mesenchymal transition and proliferation of retinal pigment epithelial cells initiated upon loss of cell-cell contact. Investig. Ophthalmol. Vis. Sci. 2010, 51, 2755–2763.
- Tamiya, S.; Kaplan, H.J. Role of epithelial-mesenchymal transition in proliferative vitreoretinopathy. Exp. Eye Res. 2016, 142, 26–31.
- Zhou, M.; Geathers, J.S.; Grillo, S.L.; Weber, S.R.; Wang, W.; Zhao, Y.; Sundstrom, J.M. Role of Epithelial-Mesenchymal Transition in Retinal Pigment Epithelium Dysfunction. Front. Cell Dev. Biol. 2020, 8, 501.
- Yan, X.; Lin, J.; Rolfs, A.; Luo, J. Differential expression of the ADAMs in developing chicken retina. Dev. Growth Differ. 2011, 3, 726–739.
- Maretzky, T.; Scholz, F.; Köten, B.; Proksch, E.; Saftig, P.; Reiss, K. ADAM10-mediated E-cadherin release is regulated by proinflammatory cytokines and modulates keratinocyte cohesion in eczematous dermatitis. J. Investig. Dermatol. 2008, 128, 1737–1746.
- Morrisey, E.E. Wnt signaling and pulmonary fibrosis. Am. J. Pathol. 2003, 162, 1393–1397.
- Kar, R.; Jha, N.K.; Jha, S.K.; Sharma, A.; Dholpuria, S.; Asthana, N.; Chaurasiya, K.; Singh, V.K.; Burgee, S.; Nand, P. A “NOTCH” Deeper into the Epithelial-To-Mesenchymal Transition (EMT) Program in Breast Cancer. Genes 2019, 10, 961.
- Van Aken, E.H.; De Wever, O.; Van Hoorde, L.; Bruyneel, E.; De Laey, J.-J.; Mareel, M.M. Invasion of retinal pigment epithelial cells: N-cadherin, hepatocyte growth factor, and focal adhesion kinase. Investig. Ophthalmol. Vis. Sci. 2003, 44, 463–472.
- Wang, Y.; Gao, J.; Zhang, D.; Zhang, J.; Ma, J.; Jiang, H. New insights into the antifibrotic effects of sorafenib on hepatic stellate cells and liver fibrosis. J. Hepatol. 2010, 53, 132–144.
- Hong, F.; Chou, H.; Fiel, M.I.; Friedman, S.L. Antifibrotic activity of sorafenib in experimental hepatic fibrosis: Refinement of inhibitory targets, dosing, and window of efficacy in vivo. Dig. Dis. Sci. 2013, 58, 257–264.
- Chen, Y.L.; Lv, J.; Ye, X.L.; Sun, M.-Y.; Xu, Q.; Liu, C.-H.; Min, L.-H.; Li, H.-P.; Liu, P.; Ding, X. Sorafenib inhibits transforming growth factor b1-mediated epithelial-mesenchymal transition and apoptosis in mouse hepatocytes. Hepatology 2011, 53, 1708–1718.
- Yang, F.; Van Meter, T.E.; Buettner, R.; Hedvat, M.; Liang, W.; Kowolik, C.M.; Mepani, N.; Mirosevich, J.; Nam, S.; Chen, M.Y.; et al. Sorafenib inhibits signal transducer and activator of transcription 3 signaling associated with growth arrest and apoptosis of medulloblastomas. Mol. Cancer Ther. 2008, 7, 3519–3526.
- Blechacz, B.R.; Smoot, R.L.; Bronk, S.F.; Werneburg, N.W.; Sirica, A.E.; Gores, G.J. Sorafenib inhibits signal transducer and activator of transcription-3 signaling in cholangiocarcinoma cells by activating the phosphatase shatter proof 2. Hepatology 2009, 50, 1861–1870.
- Chen, K.-F.; Tai, W.-T.; Liu, T.-H.; Huang, H.-P.; Lin, Y.-C.; Shiau, C.-W.; Li, P.-K.; Chen, P.-J.; Cheng, A.-L. Sorafenib overcomes TRAIL resistance of hepatocellular carcinoma cells through the inhibition of STAT3. Clin. Cancer Res. 2010, 16, 5189–5199.
- Hospital, V.; Chesneau, V.; Balogh, A.; Joulie, C.; SEIDAH, N.G.; COHEN, P.; PRAT, A. N-arginine dibasic convertase (nardilysin) isoforms are soluble dibasic-specific metalloendopeptidases that localize in the cytoplasm and at the cell surface. Biochem. J. 2000, 349, 587–597.
- Nishi, E.; Hiraoka, Y.; Yoshida, K.; Okawa, K.; Kita, T. Nardilysin enhances ectodomain shedding of heparin-binding epidermal growth factor-like growth factor through activation of tumor necrosis factor-alpha-converting enzyme. J. Biol. Chem. 2006, 281, 31164–31172.
- Palau, V.; Pascual, J.; Soler, M.J.; Riera, M. Role of ADAM17 in kidney disease. Am. J. Physiol. Renal Physiol. 2019, 317, F333–F342.
- Melenhorst, W.B.; Visser, L.; Timmer, A.; van den Heuvel, M.C.; Stegeman, C.A.; van Goor, H. ADAM17 upregulation in human renal disease: A role in modulating TGF- availability? Am. J. Physiol. Renal Physiol. 2009, 297, F781–F790.
- Johnson, S.A.; Spurney, R.F. Twenty years after ACEIs and ARBs: Emerging treatment strategies for diabetic nephropathy. Am. J. Physiol. Renal Physiol. 2015, 309, F807–F820.
- Li, J.H.; Huang, X.R.; Zhu, H.J.; Johnson, R.; Lan, H.Y. Role of TGF-beta signaling in extracellular matrix production under high glucose conditions. Kidney Int. 2003, 63, 2010–2019.
- Ford, B.M.; Eid, A.A.; Göőz, M.; Barnes, J.L.; Gorin, Y.C.; Abboud, H.E. ADAM17 mediates Nox4 expression and NADPH oxidase activity in the kidney cortex of OVE26 mice. Am. J. Physiol. Renal Physiol. 2013, 305, F323–F332.
- Uttarwar, L.; Peng, F.; Wu, D.; Kumar, S.; Gao, B.; Ingram, A.J.; Krepinsky, J.C. HB-EGF release mediates glucose-induced activation of the epidermal growth factor receptor in mesangial cells. Am. J. Physiol. Renal. Physiol. 2011, 300, F921–F931.
- Li, R.; Wang, T.; Walia, K.; Gao, B.; Krepinsky, J.C. Regulation of profibrotic responses by ADAM17 activation in high glucose requires its C-terminus and FAK. J. Cell. Sci. 2018, 131, 208629.
- Jang, H.J.; Kang, K.S.; Ko, H. Stereospecific effects of ginsenoside 20-Rg3 inhibits TGF-β1-induced epithelial-mesenchymal transition and suppresses lung cancer migration, invasion and anoikis resistance. Toxicology 2014, 322, 23–33.
- Cichon, M.A.; Radisky, D.C. ROS-induced epithelial-mesenchymal transition in mammary epithelial cells is mediated by NF-κB-dependent activation of Snail. Oncotarget 2014, 5, 2827–2838.
- Hao, Y.; Baker, D.; Ten Dijke, P. TGF-β-Mediated Epithelial-Mesenchymal Transition and Cancer Metastasis. Int. J. Mol. Sci. 2019, 20, 2767.
- Tsuchida, T.; Friedman, S.L. Mechanisms of hepatic stellate cell activation. Nat. Rev. Gastroenterol. Hepatol. 2017, 14, 397–411.
- Friedman, S.L. Hepatic stellate cells: Protean, multifunctional, and enigmatic cells of the liver. Physiol Rev. 2008, 88, 125–172.
- Amann, T.; Bataille, F.; Spruss, T.; Mühlbauer, M.; Gäbele, E.; Schölmerich, J.; Kiefer, P.; Bosserhoff, A.-K.; Hellerbrand, C. Activated hepatic stellate cells promote tumorigenicity of hepatocellular carcinoma. Cancer Sci. 2009, 100, 646–653.
- Bataller, R.; Brenner, D.A. Liver fibrosis. J. Clin. Investig. 2005, 115, 209–218.
- Giannelli, G.; Koudelkova, P.; Dituri, F.; Mikulits, W. Role of epithelial to mesenchymal transition in hepatocellular carcinoma. J. Hepatol. 2016, 65, 798–808.
- Pinzani, M. Epithelial-Mesenchymal transition in chronic liver disease: Fibrogenesis or escape from death? J. Hepatol. 2011, 55, 459–465.
- Théret, N.; Bouezzedine, F.; Azar, F.; Diab-Assaf, M.; Legagneux, V. ADAM and ADAMTS Proteins, New Players in the Regulation of Hepatocellular Carcinoma Microenvironment. Cancers 2021, 13, 1563.
- Yang, B.; Wang, C.; Xie, H.; Wang, Y.; Huang, J.; Rong, Y.; Zhang, H.; Kong, H.; Yang, Y.; Lu, Y. MicroRNA-3163 targets ADAM-17 and enhances the sensitivity of hepatocellular carcinoma cells to molecular targeted agents. Cell Death Dis. 2019, 10, 784.
- Zhang, Y.; Li, D.; Jiang, Q.; Cao, S.; Sun, H.; Chai, Y.; Li, X.; Ren, T.; Yang, R.; Feng, F.; et al. Novel ADAM-17 inhibitor ZLDI-8 enhances the in vitro and in vivo chemotherapeutic effects of sorafenib on hepatocellular carcinoma cells. Cell Death Dis. 2018, 9, 743.
- Lu, H.-Y.; Chu, H.-X.; Tan, Y.-X.; Qin, X.-C.; Liu, M.-Y.; Li, J.-D.; Ren, T.-S.; Zhang, Y.-S.; Zhao, Q.-C. Novel ADAM-17 inhibitor ZLDI-8 inhibits the metastasis of hepatocellular carcinoma by reversing epithelial-mesenchymal transition in vitro and in vivo. Life Sci. 2020, 244, 117343.
- Hong, S.W.; Hur, W.; Choi, J.E.; Kim, J.-H.; Hwang, D.; Yoon, S.K. Role of ADAM17 in invasion and migration of CD133-expressing liver cancer stem cells after irradiation. Oncotarget 2016, 7, 23482–23497.
- Li, Y.; Ren, Z.; Wang, Y.; Dang, Y.-Z.; Meng, B.-X.; Wang, G.-D.; Zhang, J.; Wu, J.; Wen, N. ADAM17 promotes cell migration and invasion through the integrin 1 pathway in hepatocellular carcinoma. Exp. Cell Res. 2018, 370, 373–382.
- Saha, S.K.; Choi, H.Y.; Yang, G.M.; Biswas, P.K.; Kim, K.; Kang, G.H.; Gil, M.; Cho, S.G. GPR50 Promotes Hepatocellular Carcinoma Progression via the Notch Signaling Pathway through Direct Interaction with ADAM17. Mol. Ther. Oncolytics 2020, 17, 332–349.
- Rajasekaran, S.; Rajaguru, P.; Gandhi, P.S.S. MicroRNAs as potential targets for progressive pulmonary fibrosis. Front. Pharmacol. 2015, 6, 254.
- Yang, G.; Lu, W.; Yu, D.; Sun, C.; Guo, J.; Li, Z.; Guan, F. Quantitative Analysis of Dierential Proteome Expression in Epithelial-to-Mesenchymal Transition of Bladder Epithelial Cells Using SILAC Method. Molecules 2016, 21, 84.
- Gooz, M. ADAM-17: The enzyme that does it all. Crit. Rev. Biochem. Mol. Biol. 2010, 45, 146–169.
- Sun, J.; Jiang, J.; Lu, K.; Chen, Q.; Tao, D.; Chen, Z. Therapeutic potential of ADAM17 modulation in gastric cancer through regulation of the EGFR and TNF-alpha signalling pathways. Mol. Cell. Biochem. 2017, 426, 17–26.
- Bell, H.L.; Gooz, M. ADAM-17 Is Activated by the Mitogenic Protein Kinase ERK in a Model of Kidney Fibrosis. Am. J. Med. Sci. 2010, 339, 105–107.
- Pruessmeyer, J.; Ludwig, A. The good, the bad and the ugly substrates for ADAM10 and ADAM17 in brain pathology, inflammation and cancer. Semin. Cell Dev. Biol. 2009, 20, 164–174.
- Kreuter, M.; Bonella, F.; Wijsenbeek, M.; Maher, T.M.; Spagnolo, P. Pharmacological Treatment of Idiopathic Pulmonary Fibrosis: Current Approaches, Unsolved Issues, and Future Perspectives. Biomed. Res. Int. 2015, 2015, 329481.
- Ikeda, Y.; Imai, Y.; Kumagai, H.; Nosaka, T.; Morikawa, Y.; Hisaoka, T.; Manabe, I.; Maemura, K.; Nakaoka, T.; Imamura, T.; et al. Vasorin, a transforming growth factor beta-binding protein expressed in vascular smooth muscle cells, modulates the arterial response to injury in vivo. Proc. Natl. Acad. Sci. USA 2004, 101, 10732–11077.
- Uhal, B.D.; Nguyen, H.; Dang, M.; Gopallawa, I.; Jiang, J.; Dang, V.; Ono, S.; Morimoto, K. Abrogation of ER stress-induced apoptosis of alveolar epithelial cells by angiotensin 1–7. Am. J. Physiol. Lung Cell Mol. Physiol. 2013, 305, L33–L41.
- Blom, I.E.; Goldschmeding, R.; Leask, A. Gene regulation of connective tissue growth factor: New targets for antifibrotic therapy? Matrix Biol. 2002, 21, 473–482.
- Mori, T.; Kawara, S.; Shinozaki, M.; Hayashi, N.; Kakinuma, T.; Igarashi, A.; Takigawa, M.; Nakanishi, T.; Takehara, K. Role and interaction of connective tissue growth factor with transforming growth factor-beta in persistent fibrosis: A mouse fibrosis model. J. Cell. Physiol. 1999, 181, 153–159.
- Sakai, N.; Tager, A.M. Fibrosis of two: Epithelial cell-fibroblast interactions in pulmonary fibrosis. Biochim. Biophys. Acta 2013, 1832, 911–921.
- Shafieian, M.; Chen, S.; Wu, S. Integrin-linked kinase mediates CTGF-induced epithelial to mesenchymal transition in alveolar type II epithelial cells. Pediatr. Res. 2015, 77, 520–527.
- Lin, C.H.; Nai, P.L.; Bien, M.Y.; Yu, C.C.; Chen, B.C. Thrombin-induced CCAAT/enhancer-binding protein beta activation and IL-8/CXCL8 expression via MEKK1, ERK, and p90 ribosomal S6 kinase 1 in lung epithelial cells. J. Immunol. 2014, 192, 338–348.
- Kim, S.G.; Lee, S.J. PI3K, RSK, and mTOR signal networks for the GST gene regulation. Toxicol. Sci. 2007, 96, 206–213.
- Ou, S.C.; Bai, K.J.; Cheng, W.H.; Chen, J.Y.; Lin, C.H.; Wen, H.C.; Chen, B.C. TGF-β Induced CTGF Expression in Human Lung Epithelial Cells through ERK, ADAM17, RSK1, and C/EBPβ Pathways. Int. J. Mol. Sci. 2020, 21, 9084.
- Weng, L.; Wang, W.; Su, X.; Huang, Y.; Su, L.; Liu, M.; Sun, Y.; Yang, B.; Zhou, H. The Effect of cAMP-PKA Activation on TGF-beta1-Induced Profibrotic Signaling. Cell. Physiol. Biochem. 2015, 36, 1911–1927.
- Kato, A.; Okura, T.; Hamada, C.; Miyoshi, S.; Katayama, H.; Higaki, J.; Ito, R. Cell stress induces upregulation of osteopontin via the ERK pathway in type II alveolar epithelial cells. PLoS ONE 2014, 9, e100106.


