Recently, new impetus has been given to research in this field, finding that connective tissue growth factor (CTGF), an immediate-early protein mediated by TGF-β, regulates the growth of fibroblasts and the secretion of ECM
[99][104]. A previous study suggested that subcutaneous co-injection of TGF-β plus CTGF induced sustained fibrosis in mice
[100][105]. In lung tissue obtained from IPF patients, an enhanced expression of both CTGF protein and mRNA was observed
[101][106], and, moreover, CTGF/integrin-linked kinase signalling mediates the activation of EMT in lung alveolar epithelial cells
[102][107]. Data recently collected revealed an unexpected role for ADAM17 in the regulation of this phenomenon, showing that TGF-β might activate ERK, ADAM17 and Ribosomal S6 kinase-1 (RSK1) signalling pathways; this activation cascade determines the phosphorylation of the enhancer-binding protein β (C/EBPβ) that binds the CTGF promoter region, leading to CTGF synthesis and expression. These investigations start from several lines of evidence of the role of RSK1 and protein kinase C (PKC) in the phosphorylation of C/EBPβ, a transcription factor that participates in the modulation of pro-inflammatory protein expression
[103][104][108,109]. Moreover, CTGF participates in the mechanism leading to TGF-induced FN expression in human lung epithelial cells
[105][110]; in an experimental model represented by TGF-β-induced renal fibrosis in mice, this mechanism was inhibited, blocking MEK activity and so attenuating CTGF expression
[106][111]; however, it remains unclear as to whether RSK1 and C/EBPβ are involved in TGF-β-induced CTGF expression in human lung epithelial cells and what is ADAM17’s role in EMT activation in the lung. Starting from the results obtained from Blom et al.
[99][104], suggesting that CTGF acts as a modulator of TGF-β-dependent fibrogenesis and EMT activation in lung epithelial cells
[99][104], studies have since progressed; it was demonstrated that TGF-β-induced CTGF expression in human lung epithelial cells provides the participation of ERK, ADAM17, RSK1 and C/EBPβ
[105][110], and both ADAM17 and CTGF seem to mediate TGF-β-induced FN expression
[105][110]. It is clear now the involvement of ADAM17 in the TGF-β-induced expression of CTGF and EMT in the lung; in fact, ADAM17 gene silencing reduced TGF-β-induced CTGF and FN expression in human alveolar basal epithelial cell line
[105][110]. Furthermore, through the use of CTGF gene knockdown, a reduced TGF-β-induced FN expression was observed, confirming the correlated importance of ADAM17 and CTGF in TGF-β-induced FN expression in human lung epithelial cells
[105][110]. Further clarifications on the molecular mechanisms involved in IPF fibrosis emerged through studying the ERK pathway activation that seems to regulate the expression of pro-fibrotic proteins in IPF such as osteopontin
[107][112]. By using a specific ERK inhibitor, TGF-β-induced CTGF expression was reduced in the alveolar basal epithelial cells to levels comparable to those obtained through the use of RSK1 gene silencing; additionally, TGF-β enhanced the phosphorylation of ERK and RSK1 that was decreased by using ERK inhibitors
[105][110]. This experimental strategy allowed for the demonstration that ERK mediates TGF-β-induced ADAM17 phosphorylation, and ADAM17 regulates TGF-β-induced RSK1 phosphorylation; overall, these results suggest that the ERK/ADAM17/RSK1 signalling pathway activation was required for TGF-β enhanced CTGF expression in the lung
[105][110].
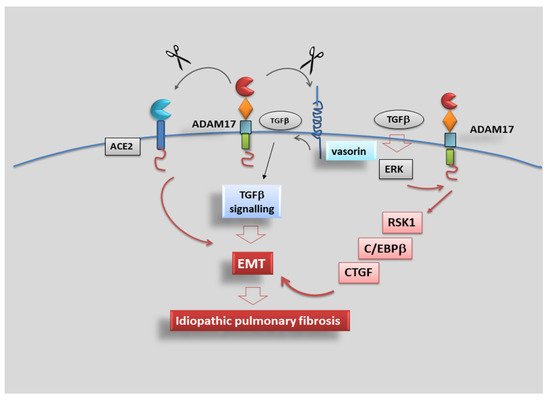
The same ERK/ADAM17/RSK1 pathway has a positive effect on C/EBPβ phosphorylation and activation, and ADAM17 plays a key role in TGF-β-induced CTGF expression and EMT through the ERK/RSK1/C/EBPβ pathway
[105][110].
In conclusion, data collected evidenced that TGF-β activates the ERK/ADAM17/RSK1/C/EBPβ signalling pathway, after which it promotes the link of C/EBPβ to the C/EBPβ site on the CTGF promoter region to regulate CTGF expression in human lung epithelial cells, revealing a signalling pathway related to ADAM17-dependent EMT and fibrosis, which may provide a new therapeutic orientation for the treatment of IPF (
Figure 5).
Figure 5. ADAM17 promotes idiopathic pulmonary fibrosis via EMT activation. Representative scheme illustrating the results of TGF-β-induced CTGF expression mediated via the ERK/ADAM17/RSK1/C/EBPβ pathway in human lung epithelial cells. TGF-β activates the ERK/ADAM17/RSK1/C/EBPβ signalling pathway, which finally leads to CTGF expression, promoting EMT in human lung epithelial cells.