Idiopathic pulmonary fibrosis (IPF) is a type of interstitial lung disease that is prevalent in elder smokers. The phases of IPF include alveolar epithelial cell damage and activation, inflammatory cell infiltration, EMT initiation and ECM protein accumulation [
96]. During the progression of IPF, most fibroblasts originate from lung epithelial cells, which undergo EMT and play a crucial role in fibrotic disease progression. The TGF-β signalling pathway has been suggested to contribute to the EMT process and produce ECM proteins, such as fibronectin (FN) [
97]. Therefore, TGF-β and EMT may be a hallmark of fibroblast activation.
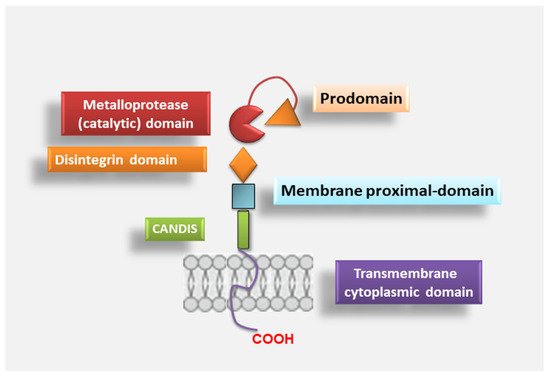
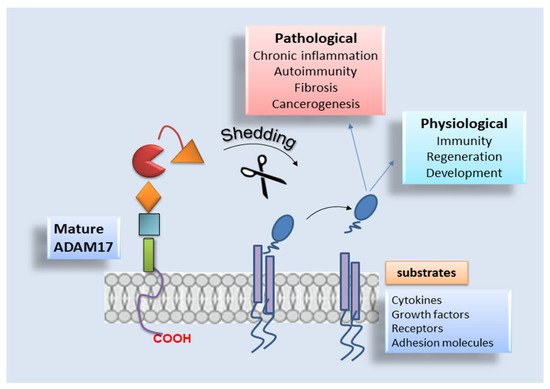
As reported above, ADAM17 is responsible for the cleavage of extracellular domains of substrate proteins [
54], thus regulating some important physiological and pathophysiological processes and the expression of membrane-bound proteins such as cytokines and growth factors [
98]. Increased ADAM17 expression is identified in several inflammatory diseases, cancers, and organ fibrotic changes, including IPF [
35,
36,
99,
100]. During the progression of chronic IPF, the volume and ventilation of the lungs are gradually decreased due to abnormal proliferation of fibroblasts through the EMT process, which causes collagen deposition and finally leads to architectural distortion [
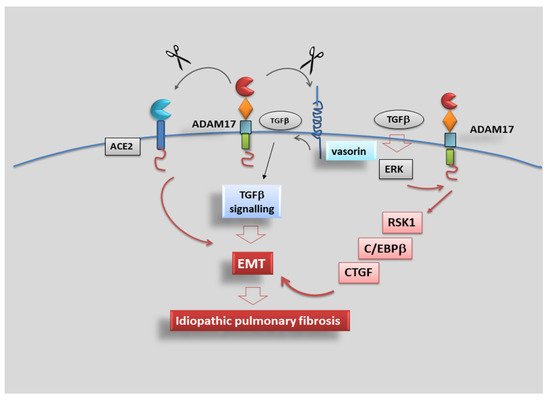
101]. A recent study demonstrated that ADAM17 regulates TGF-β-mediated EMT through the cleavage of vasorin (VSN), a type I transmembrane protein initially identified in screening to isolate novel proteins containing a signal sequence. VSN was shown to attenuate TGF-β signalling by sequestering the growth factor, and its expression was found restricted to the aorta, kidney and placenta [
102,
103]. In addition, ADAM17 is responsible for the angiotensin-converting enzyme 2 (ACE-2) ectodomain shedding occurring in lung fibrogenesis, demonstrating that ADAM17 certainly participated in IPF [
103]. However, the role of ADAM17 in TGF-β-induced EMT in IPF remains uncertain.
Recently, new impetus has been given to research in this field, finding that connective tissue growth factor (CTGF), an immediate-early protein mediated by TGF-β, regulates the growth of fibroblasts and the secretion of ECM [
104]. A previous study suggested that subcutaneous co-injection of TGF-β plus CTGF induced sustained fibrosis in mice [
105]. In lung tissue obtained from IPF patients, an enhanced expression of both CTGF protein and mRNA was observed [
106], and, moreover, CTGF/integrin-linked kinase signalling mediates the activation of EMT in lung alveolar epithelial cells [
107]. Data recently collected revealed an unexpected role for ADAM17 in the regulation of this phenomenon, showing that TGF-β might activate ERK, ADAM17 and Ribosomal S6 kinase-1 (RSK1) signalling pathways; this activation cascade determines the phosphorylation of the enhancer-binding protein β (C/EBPβ) that binds the CTGF promoter region, leading to CTGF synthesis and expression. These investigations start from several lines of evidence of the role of RSK1 and protein kinase C (PKC) in the phosphorylation of C/EBPβ, a transcription factor that participates in the modulation of pro-inflammatory protein expression [
108,
109]. Moreover, CTGF participates in the mechanism leading to TGF-induced FN expression in human lung epithelial cells [
110]; in an experimental model represented by TGF-β-induced renal fibrosis in mice, this mechanism was inhibited, blocking MEK activity and so attenuating CTGF expression [
111]; however, it remains unclear as to whether RSK1 and C/EBPβ are involved in TGF-β-induced CTGF expression in human lung epithelial cells and what is ADAM17’s role in EMT activation in the lung. Starting from the results obtained from Blom et al. [
104], suggesting that CTGF acts as a modulator of TGF-β-dependent fibrogenesis and EMT activation in lung epithelial cells [
104], studies have since progressed; it was demonstrated that TGF-β-induced CTGF expression in human lung epithelial cells provides the participation of ERK, ADAM17, RSK1 and C/EBPβ [
110], and both ADAM17 and CTGF seem to mediate TGF-β-induced FN expression [
110]. It is clear now the involvement of ADAM17 in the TGF-β-induced expression of CTGF and EMT in the lung; in fact, ADAM17 gene silencing reduced TGF-β-induced CTGF and FN expression in human alveolar basal epithelial cell line [
110]. Furthermore, through the use of CTGF gene knockdown, a reduced TGF-β-induced FN expression was observed, confirming the correlated importance of ADAM17 and CTGF in TGF-β-induced FN expression in human lung epithelial cells [
110]. Further clarifications on the molecular mechanisms involved in IPF fibrosis emerged through studying the ERK pathway activation that seems to regulate the expression of pro-fibrotic proteins in IPF such as osteopontin [
112]. By using a specific ERK inhibitor, TGF-β-induced CTGF expression was reduced in the alveolar basal epithelial cells to levels comparable to those obtained through the use of RSK1 gene silencing; additionally, TGF-β enhanced the phosphorylation of ERK and RSK1 that was decreased by using ERK inhibitors [
110]. This experimental strategy allowed for the demonstration that ERK mediates TGF-β-induced ADAM17 phosphorylation, and ADAM17 regulates TGF-β-induced RSK1 phosphorylation; overall, these results suggest that the ERK/ADAM17/RSK1 signalling pathway activation was required for TGF-β enhanced CTGF expression in the lung [
110].
In conclusion, data collected evidenced that TGF-β activates the ERK/ADAM17/RSK1/C/EBPβ signalling pathway, after which it promotes the link of C/EBPβ to the C/EBPβ site on the CTGF promoter region to regulate CTGF expression in human lung epithelial cells, revealing a signalling pathway related to ADAM17-dependent EMT and fibrosis, which may provide a new therapeutic orientation for the treatment of IPF (Figure 5).