 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Agnieszka Sliwinska | + 5130 word(s) | 5130 | 2021-07-13 10:28:38 | | | |
| 2 | Conner Chen | Meta information modification | 5130 | 2021-07-22 15:15:10 | | |
Video Upload Options
It seems that vitamin D deficiency may be one of the crucial factors responsible for increased cancer risk among T2DM patients. Vitamin D via alleviation of insulin resistance, hyperglycemia, oxidative stress and inflammation reduces diabetes driven cancer risk factors. Moreover, vitamin D strengthens the DNA repair process, and regulates apoptosis and autophagy of cancer cells as well as signaling pathways involved in tumorigenesis i.e., tumor growth factor β (TGFβ), insulin-like growth factor (IGF) and Wnt-β-Cathenin. It should also be underlined that many types of cancer cells present alterations in vitamin D metabolism and action as a result of Vitamin D Receptor (VDR) and CYP27B1 expression dysregulation.
1. Metabolic Phenotype of Vitamin D in Cancer Cells
Physiologically, VDR and CYP27B1 are expressed in numerous tissue in which they govern many functions [1]. However, in many type of cancer cells this VDR and CYP27B1-mediated regulation is disturbed which leads to disorders of vitamin D metabolism and action [2][3][4][5].
1.1. CYP27B1
It was observed that CYP27B1 expression is inversely correlated with the progression of tumors of prostate, lung, parathyroid, colon and skin [6][7][8][9][10][11][12], suggesting that local production of 1,25(OH)2D3 in CYP27B1-expressed tissues could be crucial for cancer prevention. The results of recent study have revealed that pro-inflammatory cytokines including IL-6 and TNF-α decreased CYP27B1 expression in colon cancer cells [13]. Thus, the pro-inflammatory tumor microenvironment is proposed to be a potential factor that reduces CYP27B1 level during tumor progression. The molecular mechanisms responsible for reduced CYP27B1 expression in the process of cancer progression are still not fully understood.
On the contrary, a positive association between reduced CYP27B1 expression and cancer progression was observed in thyroid cancer [14], but inconsistent results were documented for breast [15][16] and renal cancers [17][18]. Additionally, the expression of CYP27B1 in alveolar macrophages from lung cancer patients presented a positive correlation with cancer progression [19]. Notably, pro-inflammatory cytokines including IFN-γ and TNF-α, and Toll-like receptor (TLR) agonists increased CYP27B1 expression in macrophages, monocytes and dendritic cells [16][20]. These observations suggest that the pro-inflammatory tumor microenvironment may contribute to increased CYP27B1 expression in immune cells, which is inconclusive to the outcomes in colon cancer cells [13] mentioned above.
Taken together, CYP27B1 is a biomolecule that may constitute a potential target in cancer therapy. Although, the molecular mechanisms that regulate CYP27B1 expression in particular types of cancer are not fully recognized.
1.2. CYP24A1
Taking into consideration that CYP24A1 degrades both calcidiol and calcitriol, its expression might be upregulated by cancer cells and lead to reduced local concentrations of 1,25(OH)2D3 reported by Albertson et al., who found amplified CYP24A1 in breast cancer [21]. The increased expression of CYP24A1 was demonstrated to be correlated with the advanced stages of prostate, colon, lung and breast cancers, stimulating resistance to vitamin D-mediated therapy [11][15][22][23][24][25][26]. The overexpression of CYP24A1 has been also documented in numerous other types of cancer, including cervical, ovarian, squamous cell and basal cell carcinoma [27][28]. Moreover, CYP24A1 up-expression is related to poor prognosis in colon, lung and esophageal cancer [22][29][30] The oncogenic role of CYP24A1 is supported by results of studies presented that the suppression of CYP24A1 inhibited tumor growth and strengthened antitumorigenic effects of 1,25(OH)2D3 in breast and lung cancers [31][32][33]. However, opposite data have been also published for prostate cancer [24][34] and a negative correlation between expression of CYP24A1 and tumor progression has been observed in melanoma [35].
It was proposed that increased CYP24A1 expression observed in cancer cells is probably mediated via activation of VDR, because both activity and expression of VDR are downregulated in most types of cancer. Moreover, the overexpression of CYP24A1 in numerous cancer cells may not be a result of normal physiological processes mediated by calcitriol–VDR-dependent mechanisms. Firstly, it has been shown that overexpression of CYP24A1 in breast cancer is related to the amplification of chromosomal locus 20q13.2–20q13.3 comprising the CYP24A1 gene, that has been also found in other types of cancer, including colon malignancies [21][36]. The amplification of CYP24A1 was identified only in malignant, but not benign colon tumors, thus these observations suggest that CYP24A1 overexpression and inactivation of calcitriol may be a key feature of tumor cells [37]. Secondly, epigenetic modifications, namely DNA methylation of the promoter region leads to modification of CYP24A1 expression in cancer cells. It has been reported that CYP24A1 expression is negatively correlated with the methylation of the CYP24A1 promoter in prostate and lung cancer, in vivo and in vitro [29][37] Thirdly, the suppression of DNA methyltransferase (DNMT) or histone deacetylase (HDAC) elevated CYP24A1 expression in colon and lung cancer. Fourthly, post-transcriptional regulation via microRNAs is related to the CYP24A1 overexpression in cancer. The expression of CYP24A1 has been found to be inversely correlated to the expression of miR-125b in breast cancer [38], suggesting that decreased levels of miR-125b may be responsible for CYP24A1 overexpression in cancer. The results of recent study also showed that the miR-17 to -92 cluster also control CYP24A1 expression in lung cancer cells [39]. It is also known that the serine/threonine protein kinase casein kinase 2 (CK2) signaling pathway stimulates overexpression of CYP24A1 in prostate cancers. CK2 is involved in the regulation of CYP24A1 expression by 1,25(OH)2D3 and the CK2 inhibitor enhances 1,25(OH)2D3-mediated antitumor effect [40]. Moreover, CK2 overexpression has been shown in numerous cancers, including prostate, pancreatic, breast, colon and rectum, lung and bronchus cancer [41]. It should be underlined that CK2 overexpression was found to be related to poor clinical outcomes [42].
Summarizing, it seems that overexpression of CYP24A1 may lead to decreased level of calcitriol in cancer.
1.3. VDR/RXRα
Progressively reduced expression of VDR during dedifferentiation and tumor progression in many types of cancer has been observed. Moreover, a negative correlation between VDR expression and tumor malignancy has been shown during the analysis of VDR expression levels in normal, benign, and malignant tissues of ovarian, breast, skin and prostate [6][15][43][44][45][46]. Decreased expression of VDR protein has been also observed in urothelial bladder cancer and related with poor prognosis [47]. This evidence suggests that low VDR expression may be a potential early diagnostic biomarker for high-risk subjects. Several mechanisms were identified to regulate the expression of VDR. Snail1 and Snail2, members of Snail family transcriptional repressor upregulated in may cancers engaged in tumor invasion and metastasis, were revealed to bind to E-boxes in the proximal promoter region of the VDR gene leading to recruitment of co-repressors that strength the VDR transcription in breast and colon cancer cells [48][49][50]. It has been observed that the expression of H-Ras mutants in rat intestinal epithelial cells and mouse colon as well as the expression of K-Ras mutants in human colon cancer cells suppress calcitriol-mediated activation of VDR activation by inhibiting VDR transcription [51]. Moreover, decreased expression of VDR, H-Ras and K-Ras mutations in keratinocytes and human prostate epithelial cell lines are related to inhibition of VDR transcriptional activity. In turn, suppressed VDR transcriptional activity is a result of stimulation of RXR phosphorylation. RXR phosphorylation disturbs the recruitment of co-activator SRC-1 to RXR [52][53].
Epigenetic silencing of VDR has been also observed in cancer. The methylation of CpG island in the VDR promoter region has been related to decreased VDR expression in breast and colon cancer cells [54][55]. It was also observed that, DNA methyltransferase (DNMT) inhibitor stimulated VDR expression and strengthened the anti-proliferative effect of 1,25(OH)2D3 in breast cancer cells [55]. The engagement of microRNA in the control of VDR expression in cancer has also been proposed. mir-125b was demonstrated to downregulate of VDR expression and resulting resistance of melanoma cells to 1,25(OH)2D3 [56][57].
Taken together, decreased expression of VDR is a distinct feature of cancer cells and is associated with the reduced action of vitamin D.
2. Molecular Insight into Anti-Cancer Activity of Vitamin D
2.1. Anti-Inflammatory Activity of Vitamin D
Both chronic and acute hyperglycemia trigger increased level of oxidative stress, which in turn contributes to the activation of NF-kB and numerous pro-inflammatory mediators i.e., TNF-α and IL-6. The elevated level of pro-inflammatory cytokines is a key component of low grade inflammation in T2DM subjects [58]. Chronic inflammation extends inflammatory response, leading to progressive destruction and degeneration of tissues by the action of reactive oxygen species (ROS) and cytokines secreted in the site of inflammation. Thereby, chronic inflammation contributes to the initiation of tumorigenesis [59]. Vitamin D exerts anti-inflammatory effects in tumorigenesis via targeting several pathways, including prostaglandin, cyclooxygenase (COX), and mitogen activated protein kinase (MAPK) pathway.
Vitamin D is able to regulate the interaction between immune system and cancer cells resulting in the inhibition of pro-inflammatory cytokines secretion. Co-culture experiments using colon cancer cells and peripheral blood mononuclear cells (PBMCs) showed significant reduction in the secretion of pro-inflammatory cytokines by PBMCs i.e., TNF-α, IL-6 and, IL-10 after vitamin D treatment, supporting the anti-inflammatory properties of vitamin D in tumor microenvironment [60].
Nuclear factor kappa B (NF-κB) is a well-known master regulator of crosstalk between carcinogenesis and inflammation at multiple levels. Tumorous tissues are characterized by increased NF-κB activity, and the accumulation of pro-inflammatory cytokines creates the so called pro-tumorigenic microenvironment [61]. It has been documented that 1,25(OH)2D3 suppresses the NFκB signaling pathway. Calcitriol inhibits the phosphorylation of both AKT and its downstream target I kappa Bα (IκBα) via upregulation of thioesterase superfamily member 4 (THEM4) in macrophages. THEM4 is an AKT stimulator protein which upregulation results in the reduction of NF-κB and COX-2 expression [62]. Moreover, 1,25(OH)2D3 augments the stability of IκBα protein. In fibroblasts, calcitriol augmented the protein stability of IκBα. VDR physically interacts with IκB kinase β (IKKβ) to suppress NF-κB activation. VDR-IKKβ interaction blocks the formation of the IKK complex leading to the inhibition of IKKβ phosphorylation at Ser-177 and abolishing IKK activity to phosphorylate IκBα. Finally, the stabilization of IκBα suppresses the translocation of the p65/p50 complex of NFκB to the nucleus and expression of pro-inflammatory cytokines [63][64]. Together, these data define a novel mechanism of 1,25(OH)2D3–VDR mediated inhibition of NF-κB activation (presented in Figure 1).
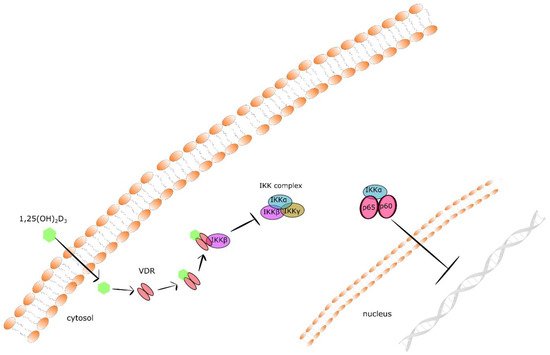
Figure 1. The mechanism of 1,25(OH)2D3–VDR mediated inhibition of NF-κB activation.
It was proposed that 1,25(OH)2D3 inhibits prostaglandin pathway engaged in pro-inflammatory responses via the suppression of the cyclooxygenase-2 and prostaglandin receptor EP2 as well as prostaglandin F receptor (FP) expression, and degradation of prostaglandins. Additionally, the upregulation of 15-hydroxyprostaglandin-dehydrogenase—NAD+-dependent degrading enzyme was observed after exposure to 1,25(OH)2D3 in prostate cancer cells [65][66]. Reduced mRNA expression of cyclooxygenase-2 and production of prostaglandin E2 have been also documented in 1,25(OH)2D3-stimulated breast cancer cells [67]. Of note, an inverse correlation between VDR expression and cyclooxygenase-2 expression has been also identified in ovarian cancer tissues and malignant breast cancer cell lines [68][69], supporting the role of 1,25(OH)2D3-VDR axis in the inhibition of cyclooxygenase expression and prostaglandins production.
P38 MAPK pathway was proposed as both a tumor suppressor and tumor promoter. Despite many studies that provided experimental findings of the antitumorigenic role of p38, many results show also that this kinase promotes cancer development via enhancing migration, survival, resistance to stress and chemotherapeutic agents in tumor cells [70]. 1,25(OH)2D3 was found to suppress the secretion of pro-inflammatory cytokines i.e., IL-6 via stimulation of MAPK phosphatase-5 expression in both normal prostate epithelial cells and prostate cancer cells. MAPK phosphatase-5 prevents phosphorylation and activation of p38 MAPK [71]. Moreover, 1,25(OH)2D3 inhibited lipopolysaccharides (LPS)-induced production of IL-6 as well as TNF-α via the activation of MAPK phosphatase-1 in murine macrophages and human monocytes [72].
Taken together, vitamin D shows anti-inflammatory properties, especially by the reduction of pro-inflammatory cytokines expression and regulation of inflammatory signaling pathways.
2.2. Antioxidant Properties of Vitamin D
Hyperglycemia, a typical sign of diabetes, leads to elevated production of reactive oxygen species (ROS), and trigger to DNA damage [73]. The major sources of free radicals in people with diabetes are as follows: increased mitochondrial leakage of superoxide anions radical from respiratory chain, glucose autooxidation, oxidative degradation of advanced glycation end-products, activation of sorbitol and hexosamine pathway [74]. The accumulation of free radicals, especially ROS and nitrogen reactive species leads, to the activation of numerous pathways that control apoptosis and differentiation of cells [75][76]. For these reasons, the maintenance of proper function of antioxidant defense systems is a key step in preventing tumor development. It has been also proposed that vitamin D may protect from DNA damage-induced by oxidative stress via the stimulation of antioxidant defenses [77]. Increased oxidative stress-induced DNA damage has been observed in colon epithelial cells of VDR-knockout mice [78]. Additionally, the supplementation of rats with calcitriol significantly decreased level of malondialdehyde—the end product of lipid peroxidation [79]. It has been also found that vitamin D supplementation reduced oxidative DNA damage in human peripheral blood lymphocytes presenting its protective role against oxidative stress-induced DNA damage in humans [80].
The protection against oxidative stress exhibited by vitamin D is related with its molecular mechanism of action that stimulates the expression of numerous enzymes participating in ROS detoxification. It was shown that 1,25(OH)2D3 stimulated the expression of superoxide dismutase 1 and 2 in prostate epithelial cells and in androgen-sensitive prostate cancer cells [81][82]. Calcitriol also induced the expression of thioredoxin reductase 1 in breast and prostate cancer cells [81][83]. Moreover, 1,25(OH)2D3 induced expression of glucose-6-phosphate dehydrogenase that is responsible for the production of NADPH for glutathione regeneration in ovarian and prostate cancer cells [84][85]. It has been also shown that NF-E2-related factor-2 (NRF2) increasing antioxidant enzymes’ expression is regulated by vitamin D [86][87]. Vitamin D has been also proposed as a regulator of cellular bioenergetics in the mitochondria in VDR-dependent molecular mechanism. Proposed mechanism is related to the upregulation of numerous molecules engaged in mitochondrial function, especially mitochondrial respiration [88][89]. It is also known that VDR is able to enter the mitochondrion by permeability transition pores [90] and governs its functions [91]. In turn, vitamin D deficiency is related to a decline in the mitochondrial respiration process as a consequence of the decreasing of proteins and nuclear mRNA molecules involved this process [88][89]. Unfortunately, the observed mechanism is still not fully explored [91]. Reduced respiration triggers a drop of mitochondrial bioenergetics leading to changes in oxidative phosphorylation, reduced ATP formation, decreased expression of complex 1 of the electron transport chain, and elevated production of ROS [92]. In turn, increasedROS level decreases the activity of the insulin signaling pathway by lowering of phosphorylation of IRS, GLUT-4 transcription, and alterations of mitochondrial activity [93][94][95]. Observed effects are supported by the findings of the study presenting that 1,25(OH)2D3/VDR signaling suppresses the process of mitochondrial respiration and differentiation of brown adipose cells [96]. It was also shown that vitamin D in VDR-mediated mechanism protected cells from the excess production of ROS that leads to cell damage [97].
Summarizing, vitamin D exhibits antioxidative properties, especially by the regulation of antioxidants’ genes expression.
2.3. DNA Repair Process
Both mitochondrial and nuclear DNA damage are a source of numerous mutations that in turn may trigger malignant transformation [98]. It has been also observed that T2DM is related not only to increased levels of oxidative DNA damage, but also to elevated susceptibility to mutagens and reduced efficiency of DNA repair [99]. Currently, a lot of research into DNA repair disorders in diabetes is conducted. It was revealed that as a result of hyperglycemia the NAD+/NADH equilibrium is shifted toward NADH. The relevant level of NAD+ is crucial for the activity of poly (adenosine diphosphate-ribose) polymerase (PARP) protein directly involved in the double strand breaks (DSB) repair process. PARP is inhibited by the NHD domain deleted in breast cancer 1 (DBC1) protein, and binding of NAD+ to the NHD domain releases PARP and allows DNA-DSB repair [100][101]. Studies performed on podocytes derived from mice models of diabetic kidney disease showed decreased expression of KAT5, that is responsible for acetylation of ataxia telangiectasia mutant (ATM), a key protein in DNA-DSB repair. The decreased activation of ATM resulting from diminished expression of KAT5 disturbs the control of checkpoints connected with cell cycle arrest, DNA repair or apoptosis [102][103]. It was also demonstrated that insulin via the inactivation of glycogen synthase kinase-3 (GSK-3β) led to impaired DNA repair. GSK-3β phosphorylates DNA repair factors such as uracil N-glycosylase (UNG2) participating in single-strand break repair associated with base excision repair (BER) and p53 binding protein 1 (53BP1) involved in repair of DSBs induced during non-homologous end joining (NHEJ) repair [104][105]. Diabetes patients present reduced expression of sirtuin 1 (SIRT1) that is responsible for deacetylation of multiple proteins, including transcription factors essential, not only for metabolic machinery, but also for DNA repair. It was found that SIRT1 deacetylated KU70 and FOXO that are recruited to DSBs sites. Thereby, SIRT1 decreased expression detected in diabetic patients significantly diminishes the efficacy of DNA repair [106][107][108].
Moreover, high concentration of glucose may suppress the expression of DNA repair protein XPD induced by insulin [109]. Thus, both increased levels of DNA damage and decreased efficacy of DNA repair are considered as cancer risk factors. Numerous studies have shown that vitamin D elevates the expression of genes engaged in DNA damage repair including p53, proliferating cell nuclear antigen (PCNA), and breast cancer 1 (BRCA1) in breast cancer cells [83], ATM, recombinant DNA repair protein (RAD50) [110], and growth arrest and DNA damage-inducible α (GADD45α) in ovarian cancer cells and squamous cell carcinoma (SCC) [111][112]. It has been also observed that vitamin D prevents the degradation of p53-binding protein 1 (53BP1) induced by cysteine proteinase Cathepsin L, that is a lysosomal endopeptidase, in breast cancer cells [113].
To conclude, vitamin D strengthens the DNA repair process by increasing the expression of genes involved in this process.
2.4. The Role of Vitamin D in Cell Cycle, Proliferation and Differentiation
It has been observed that 1,25(OH)2D3 possesses anti-proliferation and pro-differentiation activities both in normal and malignant cells [114]. The molecular mechanism responsible for the anti-proliferative activity of vitamin D is mediated by growth factor expression, numerous signaling pathways and regulation of the cell cycle. It has been demonstrated that vitamin D upregulates IGFBP3 and the cyclin-dependent kinase (CDK) inhibitors, p21 and p27, but downregulates CDK2, triggering the reduction of IGF-1- and IGF-2-induced cell proliferation, and thereby cell cycle progression [114]. Moreover, 1,25(OH)2D3 suppresses the Wnt/β-catenin signaling pathway via the inhibition of the formation of transcription factor 4-β-catenin, (TCF4-β)–catenin complexes, or the stimulation of the expression of the Wnt antagonist—Dickkopf-1 (DKK-1) [115][116]. Vitamin D-mediated activation of transcription factor, forkhead box O3/4 (FoxO3/4), has also been presented. Activated FOXO3/4 regulates the transcription of target genes engaged in cell cycle arrest and anti-proliferation i.e., GADD45A through the stimulation of its dephosphorylation and deacetylation in neuroblastoma cells [117]. Vitamin D was also observed to decrease telomerase activity. Moreover, vitamin D induces the expression of transforming growth factor β (TGFβ), its receptors, triggering the suppression of breast and colorectal cancer cell growth [118][119].
To sum up, vitamin D may also exert its anticancer activity by suppressing cell proliferation, inducing cell differentiation.
2.5. Vitamin D Is Involved in Signaling Pathways Crucial in Tumorgenesis
2.5.1. Transforming Growth Factor β (TGFβ) Signaling Pathway
TGF-β signaling plays an important role in carcinogenesis as both a tumor suppressor and an oncogene. Tumor cells escape antiproliferative effects of TGF-β via mutational inactivation or dysregulation of the expression of components in the signaling pathway. Reduced receptor function and changed ratios of the TGF-β type 1 and type 2 receptors were found in numerous tumor cells. [120]. TGFβ2 is known as a key molecule for the maintenance of tissue homeostasis. Its anti-proliferative properties have been observed in normal epithelial cells and at the early stages of carcinogenesis [121]. The TGFβ-SMAD4 signaling pathway was recognized as responsible for constraining growth and metastatic progression of prostate cancer in PTEN-null mice [122]. The treatment with 1,25(OH)2D3 or vitamin D analog elevated mRNA expression of TGFβ2 in MDA-MB-231 and MCF-7 [22], MCF10CA1a [123] as well as primary prostate cancer cells [81]. Interestingly, 1,25(OH)2D3 and its analog EB1089 induce also expression of TGFβ1 and TGFβ receptors in MCF7 breast cancer cells and 185A1 cells (immortalized mammary epithelial cells) in a mechanism requiring SMAD3 as a co-activator [118]. In turn, 1,25(OH)2D3 suppressed negative regulators of TGFβ availability, including latent TGFβ binding protein 1 (LTBP1) in OVCAR3 cells [85] andprimary prostate cancer cells [81].
1,25(OH)2D3–induced upregulation of growth differentiation factor 15 (GDF15) mRNA level has been observed in prostate cancer LNCaP cells [124]. GDF15 was demonstrated as a direct VDR target gene required for 1,25(OH)2D3–mediated growth inhibition [82]. In prostate cancer PC-3 cells, induced expression of GDF15 reduced cell proliferation, formation of soft agar clone, and xenograft tumor growth [82][91]. The influence of 1,25(OH)2D3 on the mRNA expression of other TGFβ family members, including TGFBR1, SMAD6, TGFβ1, was only observed after prolonged treatment in various cell types suggesting that the observed effect may be indirect [125][126].
Bone morphogenic proteins (BMP) are a group of growth factors that belong to the TGFβ superfamily. BMPs play an essential role in the regulation of tissue morphogenesis. In turn, BMP signaling is often disturbed in cancer, including colon cancer [127]. It was also observed 1,25(OH)2D3 or vitamin D analog regulated mRNA expression of several BMP forms i.e., BMP6 in primary prostate cancer cells [81], BMP2 and BMP6 in MCF10AT1 cells [123], and TGFβ1 and BMP2A in squamous cell carcinoma lines [125].
2.5.2. Insulin-Like Growth Factor (IGF) Signaling Pathway
Hyperinsulinemia that is associated with diabetes and obesity exerts an effect on cancer development directly, or by IGF and IGF receptors (IGFRs). It has been observed that insulin inhibits IGFBP-1 and thus elevates the free fraction of IGF-1. It is well recognized that aberrant IGF signaling focused on elevated IGF-1R activity is involved in cancer cell proliferation, migration, and invasion [128]. An indirect effect of 1,25(OH)2D3 on the growth rate of cells, as a result of interfering with the action of growth factors that induce proliferation or increase the secretion of growth factors that stimulate cell differentiation, was also proposed. IGF1-induced cell growth was suppressed by vitamin D analogs in MCF-7 cells. Moreover, observed effect was related to elevated release of IGFBP3 [129]. IGFBP3 is a molecule responsible for limiting the pro-proliferative, anti-apoptotic actions of IGF1 and IGF2 as a result of its binding to them and suppressing their ability to interact with cell surface receptors. Notably, 1,25(OH)2D3 and vitamin D analogs were found to activate the accumulation of IGFBP3 in primary prostate epithelial cell and prostate cancer cells. In turn, IGFBP3 subsequently suppresses IGF2 action [130][131]. 1,25(OH)2D3–mediated increased mRNA expression of IGFBP3 was observed in LNCaP prostate cancer cells [124] and RWPE1 cells (immortalized prostate epithelial cell line) [132]. What is more, IGFBP3 was characterized as a critical mediator of 1,25(OH)2D3–dependent inhibition of LNCaP cell growth [133]. The upregulation of many IGFBP isoforms, including IGFBP3 in prostate tissue have also been observed after a 14-day treatment with the vitamin D analog EB1089 in rats [134].
2.5.3. Wnt-β Catenin Signaling Pathway
The Wnt/β-catenin signaling pathway plays a role in numerous physiological processes, including proliferation, differentiation, apoptosis, migration, invasion and tissue homeostasis. In turn, dysregulation of the Wnt/β-catenin cascade leads to the development and progression of some tumors [135].
Vitamin D is able to arrest the cell growth as a result of disruption of β-catenin function. β-catenin is a terminal mediator of Wnt signaling. In the cytoplasm, β-catenin is found to be associated with adenomatous polyposis coli (APC) tumor suppressor protein. Induction of Wnt signaling triggers the accumulation of β-catenin and its subsequent secretion from APC. Released free β-catenin translocates to the cell nucleus where it binds with the transcription factor TCF4 on DNA strand leading to activation of transcription [136]. In turn, mutations in the APC gene disrupting APC-β-catenin interactions are often present in colon cancer [137]. Vitamin D was reported to block β-catenin-mediated gene transcription in cultured SW480-ADH [138], HT-29 and Caco-2 colon cancer cells [139] by the activation of VDR binding to β-catenin leading to the reduction of the TCF4/β-catenin transcriptional complex formation [138]. It was also observed that injections containing 1,25(OH)2D3 and 1,25(OH)2D3 analogs three times a week for 12 weeks significantly decreased polyp number in ApcMin/+ mice. Moreover, this effect was related to decreased expression of β-catenin target genes in the small intestine and colon [140]. In HEK293 kidney cells, it was shown that the AF-2 domain of VDR interacts with the C-terminus of β-catenin. [141]. 1,25(OH)2D3–induced effect can also indirectly govern β-catenin function by elevating the secretion of E-cadherin. E-cadherin is a membrane protein that binds β-catenin and prevents its nuclear accumulation. 1,25(OH)2D3 treatment was demonstrated to suppress β-catenin-induced gene transcription even in SKBR-3 cells with lack of the E-cadherin gene [141]. Therefore, these data suggest that upregulation of E-cadherin is not the only mechanism for 1,25(OH)2D3–dependent repression of β-catenin signaling. Reduced levels of nuclear β-catenin, TCF1, CD44, and c-Myc were observed in ApcMin/+ mice after 1,25(OH)2D3 injections [140]. Additionally, 1,25(OH)2D3 can also exert an effect on the expression of Wnt-signaling regulators such as Wnt activator dickkopf-4 (DKK-4). Vitamin D repressed DKK-4 [116] and increased expression of the Wnt antagonist dickkopf-1 (DKK-1) [142].
2.6. Is Vitamin D Involed in Regulation of EMT and Cancer Progression?
Physiologically, epithelial cells maintain apical-basal polarity and contact with adjacent cells via adherent junctions, tight junctions, and desmosomes [143]. After the activation of EMT, tumor epithelial cells lose their cell polarity, cell-cell adhesion and gain migratory and invasive properties, becoming mesenchymal cells [144]. Thus, EMT is a reversible process in which epithelial cells gain mesenchymal morphology and lose intercellular contacts, achieving the ability for invasion and migration [145].
1,25(OH)2D3 reduced the expression of the mesenchymal marker, vimentin, and elevated the expression of the epithelial marker, E-cadherin. In turn, it triggered to suppression of SKOV-3 cell migration and reduced TGF-β1 induced EMT. Hou et al. have shown that stimulation of SKOV-3 cells by TGF-β1 leads to tumor progression in advanced stages via numerous mechanisms including EMT [145]. Moreover, the results of in vivo and in vitro studies have suggested that 1,25(OH)2D3 and VDR inhibited the spread of ovarian cancer [146]. It has been also found that 1,25(OH)2D3 delayed malignant transformation by reducing the expression of β-catenin and elevating the expression of E-cadherin in mouse ovarian surface epithelial cells [147]. The results of animal studies have demonstrated that the exposure of ovarian cancer cells to vitamin D3 before the inoculation to immunodeficient mice reduced the potential of the cells to metastasize into lung, liver and bone marrow [148].
DEAD (Asp-Glu-Ala-Asp)-box helicase 4 (DDX4) has been recognized as another molecular target for calcitriol. The exposure to vitamin D decreased the expression of DDX which suppressed the invasion of ovarian cancer cells [149]. Interestingly, microarray studies have revealed a number of target genes engaged in tumor growth and progression mediated by 1,25(OH)2D3. It was also observed that calcitriol downregulates growth-promoting chemokines IL-8, Growth Regulated Protein-β (GRO-β), and GRO-γ [150].
Taken together, vitamin D may inhibit metastasis, especially by decreasing expression of β-cathenin and increasing expression of E-cadherin.
2.7. How Does Vitamin D Regulate Apoptosis and Autophagy of Cancer Cells?
It is known that vitamin D induces apoptosis of cancer cells via the downregulation of the anti-apoptotic proteins, Bcl-2 and Bcl-XL, and the upregulation of pro-apoptotic proteins, Bax, Bak, and Bad [151]. Moreover, the stimulation of apoptosis by increased expression of other pro-apoptotic proteins, including death-associated protein (DAP-3), G0-G1 switch 2 (GOS2), Fas-associated death domain (FADD), and caspases has been recently documented [83][126]. Calcitriol also suppresses AKT-mediated anti-apoptotic signaling pathway via upregulation of phosphatase and tensin homolog (PTEN), considered as a tumor suppressor [152]. Vitamin D can also recruit Ca2+-dependent apoptotic effectors including Ca2+-dependent μ-calpain and Ca2+/calpain-dependent caspase-12 [153].
Autophagy plays an important role in both cell survival and apoptosis-independent cell death. An ample evidence suggest that vitamin D is able to switch the mode of autophagy from survival to death in cancer cells [154][155]. Calcitriol-stimulated autophagy was associated with increased expression of beclin-1. The letter interacts with either BCL-2 or PI3K class III, playing a crucial role in the regulation of both autophagy and cell death [156]. Additionally, vitamin D-induced autophagy is a result of the stimulation of the expression of DNA damage inducible transcript 4 (DDIT4) and DNA damage response 1 (REDD1). REDD1 is known as an inhibitor of mechanistic target of rapamycin complex 1 (mTORC1) that suppresses autophagy [157][158].
Summarizing, vitamin D may induce apoptosis of cancer cells and stimulates autophagy.
2.8. The Role of Vitamin D in Angiogenesis
Blood vessels are necessary to transport oxygen and nutrients for growth and metastasis of cancer cells. Growing cancerous tissue secretes numerous proteins, including EGF, estrogen, basic and acidic FGF, IL-8, prostaglandin E1 and E2, TNF-α, and VEGF. These molecules may activate endothelial cell growth and motility when the production of anti-angiogenic factors is decreased [159].
1,25(OH)2D3 has been found to have an antiangiogenic effect by modulating the hypoxia-inducible factor 1 (HIF-1) pathway in human cancer cells. Hypoxia is the main trigger of angiogenesis in tumors. HIF-1 is a key transcription factor regulating angiogenesis. It has been documented that 1,25(OH)2D3 decreases the expression of the HIF-1α subunit, VEGF and inhibits cancer cell proliferation under hypoxic conditions [160]. The antiangiogenic effect of 1,25(OH)2D3 on tumor endothelial cells may also be VDR mediated. In VDR knockout animals, elevated vascular volume and reduced number of pericytes responsible for regulation of the proliferation of endothelial cells was observed [161].
3. Vitamin D in Cancer Prevention among Diabetes Patients
Diabetes and cancer are common chronic diseases, which frequently co-exist. Grow-ing body of evidence shows that patients with diabetes are more susceptible to the development of different cancers. The causative factors of this increased coexistence are not fully recognized. It is believed that shared pathophysiology and/or environmental risk factors may be responsible for the excess cancer risk in diabetic patients. Numerous evidence which includes epidemiological, experimental and clinical studies suggests that both cancer development and T2DM development are increased in subjects with inadequate vitamin D levels. Therefore, it can be assumed that in diabetic patients with vitamin D deficiency, the risk of cancer development will be accumulated. Taking into account the pleiotropic effect of vitamin D, especially engagement in insulin synthesis and secretion, immune response, regulation of calcium intracellular level, and response to insulin, its deficiency contributes to the intensification of typical symptoms of diabetes, such as insulin resistance, hyperinsulinism, hyperglycemia and low grade chronic inflammation. Thus, the altogether disorders accompanying diabetes create a microenvironment leading to the development of cancer, and vitamin D deficiency exacerbates their intensity. Most of the results of clinical trials involving patients suffering from T2DM show that supplementation with vitamin D improves the level of metabolic parameters associated with insulin resistance, hyperinsulinemia, hyperglycemia and low grade chronic inflammation. However, there are no clinical trials evaluating the impact of vitamin D supplementation on cancer risk among patients suffering from diabetes. Only, in the study by Wang et al. that aimed to determine the association between serum 25(OH)D concentrations and can-cer-specific mortality in 1188 older post-menopausal women, we found that diabetes did not significantly increase cancer mortality with a vitamin D cutoff of 64 nmol/L (25.6 ng/mL) [162]. Thus, there is a pressing need for randomized clinical trials to clarify whether vitamin D deficiency may be another factor responsible for increased risk of cancer in T2DM patients, and whether the use of the vitamin by patients with diabetes may decrease cancer risk.
References
- Bikle, D.D. Clinical Counterpoint: Vitamin D: New Actions, New Analogs, New Therapeutic Potential. Endocr. Rev. 1992, 13, 765–784.
- Giammanco, M.; Di Majo, D.; La Guardia, M.; Aiello, S.; Crescimannno, M.; Flandina, C.; Tumminello, F.M.; Leto, G. Vitamin D in Cancer Chemoprevention. Pharm. Biol. 2015, 53, 1399–1434.
- Bikle, D.D. Extraskeletal Actions of Vitamin D. Ann. N. Y. Acad. Sci. 2016, 1376, 29–52.
- Fleet, J.C.; DeSmet, M.; Johnson, R.; Li, Y. Vitamin D and Cancer: A Review of Molecular Mechanisms. Biochem. J. 2012, 441, 61–76.
- Bikle, D.D. Vitamin D Metabolism, Mechanism of Action, and Clinical Applications. Chem. Biol. 2014, 21, 319–329.
- Brożyna, A.A.; Jozwicki, W.; Janjetovic, Z.; Slominski, A.T. Expression of Vitamin D Receptor (VDR) Decreases during Progression of Pigmented Skin Lesions. Hum. Pathol. 2011, 42, 618–631.
- Segersten, U.; Correa, P.; Hewison, M.; Hellman, P.; Dralle, H.; Carling, T.; Akerström, G.; Westin, G. 25-Hydroxyvitamin D(3)-1alpha-Hydroxylase Expression in Normal and Pathological Parathyroid Glands. J. Clin. Endocrinol. Metab. 2002, 87, 2967–2972.
- Chen, T.C.; Holick, M.F.; Lokeshwar, B.L.; Burnstein, K.L.; Schwartz, G.G. Evaluation of Vitamin D Analogs as Therapeutic Agents for Prostate Cancer. Recent Results Cancer Res. Fortschr. Krebsforsch. Progres Dans Rech. Sur Cancer 2003, 164, 273–288.
- Hansdottir, S.; Monick, M.M.; Hinde, S.L.; Lovan, N.; Look, D.C.; Hunninghake, G.W. Respiratory Epithelial Cells Convert Inactive Vitamin D to Its Active Form: Potential Effects on Host Defense. J. Immunol. 2008, 181, 7090–7099.
- Hsu, J.-W.; Yasmin-Karim, S.; King, M.R.; Wojciechowski, J.C.; Mickelsen, D.; Blair, M.L.; Ting, H.-J.; Ma, W.-L.; Lee, Y.-F. Suppression of Prostate Cancer Cell Rolling and Adhesion to Endothelium by 1α,25-Dihydroxyvitamin D3. Am. J. Pathol. 2011, 178, 872–880.
- Matusiak, D.; Benya, R.V. CYP27A1 and CYP24 Expression as a Function of Malignant Transformation in the Colon. J. Histochem. Cytochem. Off. J. Histochem. Soc. 2007, 55, 1257–1264.
- Mawer, E.B.; Hayes, M.E.; Heys, S.E.; Davies, M.; White, A.; Stewart, M.F.; Smith, G.N. Constitutive Synthesis of 1,25-Dihydroxyvitamin D3 by a Human Small Cell Lung Cancer Cell Line. J. Clin. Endocrinol. Metab. 1994, 79, 554–560.
- Hummel, D.M.; Fetahu, I.S.; Gröschel, C.; Manhardt, T.; Kállay, E. Role of Proinflammatory Cytokines on Expression of Vitamin D Metabolism and Target Genes in Colon Cancer Cells. J. Steroid Biochem. Mol. Biol. 2014, 144, 91–95.
- Clinckspoor, I.; Hauben, E.; Verlinden, L.; Van den Bruel, A.; Vanwalleghem, L.; Vander Poorten, V.; Delaere, P.; Mathieu, C.; Verstuyf, A.; Decallonne, B. Altered Expression of Key Players in Vitamin D Metabolism and Signaling in Malignant and Benign Thyroid Tumors. J. Histochem. Cytochem. 2012, 60, 502–511.
- Lopes, N.; Sousa, B.; Martins, D.; Gomes, M.; Vieira, D.; Veronese, L.A.; Milanezi, F.; Paredes, J.; Costa, J.L.; Schmitt, F. Alterations in Vitamin D Signalling and Metabolic Pathways in Breast Cancer Progression: A Study of VDR, CYP27B1 and CYP24A1 Expression in Benign and Malignant Breast Lesions Vitamin D Pathways Unbalanced in Breast Lesions. BMC Cancer 2010, 10, 483.
- Lopes, N.; Carvalho, J.; Durães, C.; Sousa, B.; Gomes, M.; Costa, J.L.; Oliveira, C.; Paredes, J.; Schmitt, F. 1Alpha,25-Dihydroxyvitamin D3 Induces de Novo E-Cadherin Expression in Triple-Negative Breast Cancer Cells by CDH1-Promoter Demethylation. Anticancer Res. 2012, 32, 249–257.
- Urbschat, A.; Paulus, P.; von Quernheim, Q.F.; Brück, P.; Badenhoop, K.; Zeuzem, S.; Ramos-Lopez, E. Vitamin D Hydroxylases CYP2R1, CYP27B1 and CYP24A1 in Renal Cell Carcinoma. Eur. J. Clin. Investig. 2013, 43, 1282–1290.
- Blomberg Jensen, M.; Andersen, C.B.; Nielsen, J.E.; Bagi, P.; Jørgensen, A.; Juul, A.; Leffers, H. Expression of the Vitamin D Receptor, 25-Hydroxylases, 1alpha-Hydroxylase and 24-Hydroxylase in the Human Kidney and Renal Clear Cell Cancer. J. Steroid Biochem. Mol. Biol. 2010, 121, 376–382.
- Yokomura, K.; Suda, T.; Sasaki, S.; Inui, N.; Chida, K.; Nakamura, H. Increased Expression of the 25-Hydroxyvitamin D(3)-1alpha-Hydroxylase Gene in Alveolar Macrophages of Patients with Lung Cancer. J. Clin. Endocrinol. Metab. 2003, 88, 5704–5709.
- Széles, L.; Keresztes, G.; Töröcsik, D.; Balajthy, Z.; Krenács, L.; Póliska, S.; Steinmeyer, A.; Zuegel, U.; Pruenster, M.; Rot, A.; et al. 1,25-Dihydroxyvitamin D3 Is an Autonomous Regulator of the Transcriptional Changes Leading to a Tolerogenic Dendritic Cell Phenotype. J. Immunol. 2009, 182, 2074–2083.
- Albertson, D.G.; Ylstra, B.; Segraves, R.; Collins, C.; Dairkee, S.H.; Kowbel, D.; Kuo, W.L.; Gray, J.W.; Pinkel, D. Quantitative Mapping of Amplicon Structure by Array CGH Identifies CYP24 as a Candidate Oncogene. Nat. Genet. 2000, 25, 144–146.
- Chen, G.; Kim, S.H.; King, A.N.; Zhao, L.; Simpson, R.U.; Christensen, P.J.; Wang, Z.; Thomas, D.G.; Giordano, T.J.; Lin, L.; et al. CYP24A1 Is an Independent Prognostic Marker of Survival in Patients with Lung Adenocarcinoma. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2011, 17, 817–826.
- Cross, H.S.; Bises, G.; Lechner, D.; Manhardt, T.; Kállay, E. The Vitamin D Endocrine System of the Gut—Its Possible Role in Colorectal Cancer Prevention. J. Steroid Biochem. Mol. Biol. 2005, 97, 121–128.
- Tannour-Louet, M.; Lewis, S.K.; Louet, J.-F.; Stewart, J.; Addai, J.B.; Sahin, A.; Vangapandu, H.V.; Lewis, A.L.; Dittmar, K.; Pautler, R.G.; et al. Increased Expression of CYP24A1 Correlates with Advanced Stages of Prostate Cancer and Can Cause Resistance to Vitamin D3-Based Therapies. FASEB J. 2014, 28, 364–372.
- Brozek, W.; Manhardt, T.; Kállay, E.; Peterlik, M.; Cross, H.S. Relative Expression of Vitamin D Hydroxylases, CYP27B1 and CYP24A1, and of Cyclooxygenase-2 and Heterogeneity of Human Colorectal Cancer in Relation to Age, Gender, Tumor Location, and Malignancy: Results from Factor and Cluster Analysis. Cancers 2012, 4, 763–776.
- Horváth, H.C.; Lakatos, P.; Kósa, J.P.; Bácsi, K.; Borka, K.; Bises, G.; Nittke, T.; Hershberger, P.A.; Speer, G.; Kállay, E. The Candidate Oncogene CYP24A1: A Potential Biomarker for Colorectal Tumorigenesis. J. Histochem. Cytochem. 2010, 58, 277–285.
- Mitschele, T.; Diesel, B.; Friedrich, M.; Meineke, V.; Maas, R.M.; Gärtner, B.C.; Kamradt, J.; Meese, E.; Tilgen, W.; Reichrath, J. Analysis of the Vitamin D System in Basal Cell Carcinomas (BCCs). Lab. Investig. J. Tech. Methods Pathol. 2004, 84, 693–702.
- Anderson, M.G.; Nakane, M.; Ruan, X.; Kroeger, P.E.; Wu-Wong, J.R. Expression of VDR and CYP24A1 MRNA in Human Tumors. Cancer Chemother. Pharmacol. 2006, 57, 234–240.
- Mimori, K.; Tanaka, Y.; Yoshinaga, K.; Masuda, T.; Yamashita, K.; Okamoto, M.; Inoue, H.; Mori, M. Clinical Significance of the Overexpression of the Candidate Oncogene CYP24 in Esophageal Cancer. Ann. Oncol. Off. J. Eur. Soc. Med. Oncol. 2004, 15, 236–241.
- Sun, H.; Wang, C.; Hao, M.; Sun, R.; Wang, Y.; Liu, T.; Cong, X.; Liu, Y. CYP24A1 Is a Potential Biomarker for the Progression and Prognosis of Human Colorectal Cancer. Hum. Pathol. 2016, 50, 101–108.
- Osanai, M.; Lee, G.-H. CYP24A1-Induced Vitamin D Insufficiency Promotes Breast Cancer Growth. Oncol. Rep. 2016, 36, 2755–2762.
- Shiratsuchi, H.; Wang, Z.; Chen, G.; Ray, P.; Lin, J.; Zhang, Z.; Zhao, L.; Beer, D.; Ray, D.; Ramnath, N. Oncogenic Potential of CYP24A1 in Lung Adenocarcinoma. J. Thorac. Oncol. Off. Publ. Int. Assoc. Study Lung Cancer 2017, 12, 269–280.
- Muindi, J.R.; Yu, W.-D.; Ma, Y.; Engler, K.L.; Kong, R.-X.; Trump, D.L.; Johnson, C.S. CYP24A1 Inhibition Enhances the Antitumor Activity of Calcitriol. Endocrinology 2010, 151, 4301–4312.
- Luo, W.; Karpf, A.R.; Deeb, K.K.; Muindi, J.R.; Morrison, C.D.; Johnson, C.S.; Trump, D.L. Epigenetic Regulation of Vitamin D 24-Hydroxylase/CYP24A1 in Human Prostate Cancer. Cancer Res. 2010, 70, 5953–5962.
- Brożyna, A.A.; Jochymski, C.; Janjetovic, Z.; Jóźwicki, W.; Tuckey, R.C.; Slominski, A.T. CYP24A1 Expression Inversely Correlates with Melanoma Progression: Clinic-Pathological Studies. Int. J. Mol. Sci. 2014, 15, 19000–19017.
- Höbaus, J.; Hummel, D.M.; Thiem, U.; Fetahu, I.S.; Aggarwal, A.; Müllauer, L.; Heller, G.; Egger, G.; Mesteri, I.; Baumgartner-Parzer, S.; et al. Increased Copy-Number and Not DNA Hypomethylation Causes Overexpression of the Candidate Proto-Oncogene CYP24A1 in Colorectal Cancer. Int. J. Cancer J. Int. Cancer 2013, 133, 1380–1388.
- Meijer, G.A.; Hermsen, M.A.; Baak, J.P.; van Diest, P.J.; Meuwissen, S.G.; Beliën, J.A.; Hoovers, J.M.; Joenje, H.; Snijders, P.J.; Walboomers, J.M. Progression from Colorectal Adenoma to Carcinoma Is Associated with Non-Random Chromosomal Gains as Detected by Comparative Genomic Hybridisation. J. Clin. Pathol. 1998, 51, 901–909.
- Komagata, S.; Nakajima, M.; Takagi, S.; Mohri, T.; Taniya, T.; Yokoi, T. Human CYP24 Catalyzing the Inactivation of Calcitriol Is Post-Transcriptionally Regulated by MiR-125b. Mol. Pharmacol. 2009, 76, 702–709.
- Borkowski, R.; Du, L.; Zhao, Z.; McMillan, E.; Kosti, A.; Yang, C.-R.; Suraokar, M.; Wistuba, I.I.; Gazdar, A.F.; Minna, J.D.; et al. Genetic Mutation of P53 and Suppression of the MiR-17~92 Cluster Are Synthetic Lethal in Non-Small Cell Lung Cancer Due to Upregulation of Vitamin D Signaling. Cancer Res. 2015, 75, 666–675.
- Luo, W.; Yu, W.-D.; Ma, Y.; Chernov, M.; Trump, D.L.; Johnson, C.S. Inhibition of Protein Kinase CK2 Reduces CYP24A1 Expression and Enhances 1,25-Dihydroxyvitamin D3 Anti-Tumor Activity in Human Prostate Cancer Cells. Cancer Res. 2013, 73, 2289–2297.
- Chua, M.M.J.; Ortega, C.E.; Sheikh, A.; Lee, M.; Abdul-Rassoul, H.; Hartshorn, K.L.; Dominguez, I. CK2 in Cancer: Cellular and Biochemical Mechanisms and Potential Therapeutic Target. Pharmaceuticals 2017, 10, 18.
- Trembley, J.H.; Chen, Z.; Unger, G.; Slaton, J.; Kren, B.T.; Van Waes, C.; Ahmed, K. Emergence of Protein Kinase CK2 as a Key Target in Cancer Therapy. BioFactors Oxf. Engl. 2010, 36, 187–195.
- Hendrickson, W.K.; Flavin, R.; Kasperzyk, J.L.; Fiorentino, M.; Fang, F.; Lis, R.; Fiore, C.; Penney, K.L.; Ma, J.; Kantoff, P.W.; et al. Vitamin D Receptor Protein Expression in Tumor Tissue and Prostate Cancer Progression. J. Clin. Oncol. 2011, 29, 2378–2385.
- Thill, M.; Fischer, D.; Kelling, K.; Hoellen, F.; Dittmer, C.; Hornemann, A.; Salehin, D.; Diedrich, K.; Friedrich, M.; Becker, S. Expression of Vitamin D Receptor (VDR), Cyclooxygenase-2 (COX-2) and 15-Hydroxyprostaglandin Dehydrogenase (15-PGDH) in Benign and Malignant Ovarian Tissue and 25-Hydroxycholecalciferol (25(OH2)D3) and Prostaglandin E2 (PGE2) Serum Level in Ovarian Cancer Patients. J. Steroid Biochem. Mol. Biol. 2010, 121, 387–390.
- Al-Azhri, J.; Zhang, Y.; Bshara, W.; Zirpoli, G.; McCann, S.E.; Khoury, T.; Morrison, C.D.; Edge, S.B.; Ambrosone, C.B.; Yao, S. Tumor Expression of Vitamin D Receptor and Breast Cancer Histopathological Characteristics and Prognosis. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2017, 23, 97–103.
- Zhang, Y.; Guo, Q.; Zhang, Z.; Bai, N.; Liu, Z.; Xiong, M.; Wei, Y.; Xiang, R.; Tan, X. VDR Status Arbitrates the Prometastatic Effects of Tumor-Associated Macrophages. Mol. Cancer Res. MCR 2014, 12, 1181–1191.
- Jóźwicki, W.; Brożyna, A.A.; Siekiera, J.; Slominski, A.T. Expression of Vitamin D Receptor (VDR) Positively Correlates with Survival of Urothelial Bladder Cancer Patients. Int. J. Mol. Sci. 2015, 16, 24369–24386.
- Pálmer, H.G.; Larriba, M.J.; García, J.M.; Ordóñez-Morán, P.; Peña, C.; Peiró, S.; Puig, I.; Rodríguez, R.; de la Fuente, R.; Bernad, A.; et al. The Transcription Factor SNAIL Represses Vitamin D Receptor Expression and Responsiveness in Human Colon Cancer. Nat. Med. 2004, 10, 917–919.
- Mittal, M.K.; Myers, J.N.; Misra, S.; Bailey, C.K.; Chaudhuri, G. In Vivo Binding to and Functional Repression of the VDR Gene Promoter by SLUG in Human Breast Cells. Biochem. Biophys. Res. Commun. 2008, 372, 30–34.
- Peña, C.; García, J.M.; Silva, J.; García, V.; Rodríguez, R.; Alonso, I.; Millán, I.; Salas, C.; de Herreros, A.G.; Muñoz, A.; et al. E-Cadherin and Vitamin D Receptor Regulation by SNAIL and ZEB1 in Colon Cancer: Clinicopathological Correlations. Hum. Mol. Genet. 2005, 14, 3361–3370.
- DeSmet, M.L.; Fleet, J.C. Constitutively Active RAS Signaling Reduces 1,25 Dihydroxyvitamin D-Mediated Gene Transcription in Intestinal Epithelial Cells by Reducing Vitamin D Receptor Expression. J. Steroid Biochem. Mol. Biol. 2017, 173, 194–201.
- Solomon, C.; White, J.H.; Kremer, R. Mitogen-Activated Protein Kinase Inhibits 1,25-Dihydroxyvitamin D3–Dependent Signal Transduction by Phosphorylating Human Retinoid X Receptor α. J. Clin. Investig. 1999, 103, 1729–1735.
- Zhang, Z.; Kovalenko, P.; Cui, M.; DeSmet, M.; Clinton, S.K.; Fleet, J.C. Constitutive Activation of the Mitogen Activated Protein Kinase Pathway Impairs Vitamin D Signaling in Human Prostate Epithelial Cells. J. Cell. Physiol. 2010, 224, 433–442.
- Liel, Y.; Shany, S.; Smirnoff, P.; Schwartz, B. Estrogen Increases 1,25-Dihydroxyvitamin D Receptors Expression and Bioresponse in the Rat Duodenal Mucosa. Endocrinology 1999, 140, 280–285.
- Marik, R.; Fackler, M.; Gabrielson, E.; Zeiger, M.A.; Sukumar, S.; Stearns, V.; Umbricht, C.B. DNA Methylation-Related Vitamin D Receptor Insensitivity in Breast Cancer. Cancer Biol. Ther. 2010, 10, 44–53.
- Essa, S.; Denzer, N.; Mahlknecht, U.; Klein, R.; Collnot, E.M.; Tilgen, W.; Reichrath, J. VDR MicroRNA Expression and Epigenetic Silencing of Vitamin D Signaling in Melanoma Cells. J. Steroid Biochem. Mol. Biol. 2010, 121, 110–113.
- Mohri, T.; Nakajima, M.; Takagi, S.; Komagata, S.; Yokoi, T. MicroRNA Regulates Human Vitamin D Receptor. Int. J. Cancer 2009, 125, 1328–1333.
- Tsuji, S.; Kawai, N.; Tsujii, M.; Kawano, S.; Hori, M. Review Article: Inflammation-Related Promotion of Gastrointestinal Carcinogenesis—A Perigenetic Pathway. Aliment. Pharmacol. Ther. 2003, 18 (Suppl. 1), 82–89.
- Mantovani, A.; Allavena, P.; Sica, A.; Balkwill, F. Cancer-Related Inflammation. Nature 2008, 454, 436–444.
- Bessler, H.; Djaldetti, M. 1α,25-Dihydroxyvitamin D3 Modulates the Interaction between Immune and Colon Cancer Cells. Biomed. Pharmacother. Biomed. Pharmacother. 2012, 66, 428–432.
- Xia, Y.; Shen, S.; Verma, I.M. NF-ΚB, an Active Player in Human Cancers. Cancer Immunol. Res. 2014, 2, 823–830.
- Wang, Q.; He, Y.; Shen, Y.; Zhang, Q.; Chen, D.; Zuo, C.; Qin, J.; Wang, H.; Wang, J.; Yu, Y. Vitamin D Inhibits COX-2 Expression and Inflammatory Response by Targeting Thioesterase Superfamily Member 4. J. Biol. Chem. 2014, 289, 11681–11694.
- Sun, J.; Kong, J.; Duan, Y.; Szeto, F.L.; Liao, A.; Madara, J.L.; Li, Y.C. Increased NF-KappaB Activity in Fibroblasts Lacking the Vitamin D Receptor. Am. J. Physiol. Endocrinol. Metab. 2006, 291, E315–E322.
- Chen, Y.; Zhang, J.; Ge, X.; Du, J.; Deb, D.K.; Li, Y.C. Vitamin D Receptor Inhibits Nuclear Factor ΚB Activation by Interacting with IκB Kinase β Protein. J. Biol. Chem. 2013, 288, 19450–19458.
- Krishnan, A.V.; Feldman, D. Molecular Pathways Mediating the Anti-Inflammatory Effects of Calcitriol: Implications for Prostate Cancer Chemoprevention and Treatment. Endocr. Relat. Cancer 2010, 17, R19–R38.
- Moreno, J.; Krishnan, A.V.; Swami, S.; Nonn, L.; Peehl, D.M.; Feldman, D. Regulation of Prostaglandin Metabolism by Calcitriol Attenuates Growth Stimulation in Prostate Cancer Cells. Cancer Res. 2005, 65, 7917–7925.
- Yuan, L.; Jiang, R.; Yang, Y.; Ding, S.; Deng, H. 1,25-Dihydroxyvitamin D3 Inhibits Growth of the Breast Cancer Cell Line MCF-7 and Downregulates Cytochrome P4501B1 through the COX-2/PGE2 Pathway. Oncol. Rep. 2012, 28, 2131–2137.
- Cordes, T.; Hoellen, F.; Dittmer, C.; Salehin, D.; Kümmel, S.; Friedrich, M.; Köster, F.; Becker, S.; Diedrich, K.; Thill, M. Correlation of Prostaglandin Metabolizing Enzymes and Serum PGE2 Levels with Vitamin D Receptor and Serum 25(OH)2D3 Levels in Breast and Ovarian Cancer. Anticancer Res. 2012, 32, 351–357.
- Thill, M.; Hoellen, F.; Becker, S.; Dittmer, C.; Fischer, D.; Kümmel, S.; Salehin, D.; Friedrich, M.; Köster, F.; Diedrich, K.; et al. Expression of Prostaglandin- and Vitamin D-Metabolising Enzymes in Benign and Malignant Breast Cells. Anticancer Res. 2012, 32, 367–372.
- Martínez-Limón, A.; Joaquin, M.; Caballero, M.; Posas, F.; de Nadal, E. The P38 Pathway: From Biology to Cancer Therapy. Int. J. Mol. Sci. 2020, 21, 1913.
- Nonn, L.; Peng, L.; Feldman, D.; Peehl, D.M. Inhibition of P38 by Vitamin D Reduces Interleukin-6 Production in Normal Prostate Cells via Mitogen-Activated Protein Kinase Phosphatase 5: Implications for Prostate Cancer Prevention by Vitamin D. Cancer Res. 2006, 66, 4516–4524.
- Zhang, Y.; Leung, D.Y.M.; Richers, B.N.; Liu, Y.; Remigio, L.K.; Riches, D.W.; Goleva, E. Vitamin D Inhibits Monocyte/Macrophage Proinflammatory Cytokine Production by Targeting MAPK Phosphatase-1. J. Immunol. 2012, 188, 2127–2135.
- Dandona, P.; Thusu, K.; Cook, S.; Snyder, B.; Makowski, J.; Armstrong, D.; Nicotera, T. Oxidative Damage to DNA in Diabetes Mellitus. Lancet 1996, 347, 444–445.
- Caimi, G.; Carollo, C.; Lo Presti, R. Diabetes Mellitus: Oxidative Stress and Wine. Curr. Med. Res. Opin. 2003, 19, 581–586.
- Yakes, F.M.; Van Houten, B. Mitochondrial DNA Damage Is More Extensive and Persists Longer than Nuclear DNA Damage in Human Cells Following Oxidative Stress. Proc. Natl. Acad. Sci. USA 1997, 94, 514–519.
- Bandy, B.; Davison, A.J. Mitochondrial Mutations May Increase Oxidative Stress: Implications for Carcinogenesis and Aging? Free Radic. Biol. Med. 1990, 8, 523–539.
- Nair-Shalliker, V.; Armstrong, B.K.; Fenech, M. Does Vitamin D Protect against DNA Damage? Mutat. Res. 2012, 733, 50–57.
- Kállay, E.; Bareis, P.; Bajna, E.; Kriwanek, S.; Bonner, E.; Toyokuni, S.; Cross, H.S. Vitamin D Receptor Activity and Prevention of Colonic Hyperproliferation and Oxidative Stress. Food Chem. Toxicol. Int. J. Publ. Br. Ind. Biol. Res. Assoc. 2002, 40, 1191–1196.
- Banakar, M.C.; Paramasivan, S.K.; Chattopadhyay, M.B.; Datta, S.; Chakraborty, P.; Chatterjee, M.; Kannan, K.; Thygarajan, E. 1alpha, 25-Dihydroxyvitamin D3 Prevents DNA Damage and Restores Antioxidant Enzymes in Rat Hepatocarcinogenesis Induced by Diethylnitrosamine and Promoted by Phenobarbital. World J. Gastroenterol. 2004, 10, 1268–1275.
- Fedirko, V.; Bostick, R.M.; Long, Q.; Flanders, W.D.; McCullough, M.L.; Sidelnikov, E.; Daniel, C.R.; Rutherford, R.E.; Shaukat, A. Effects of Supplemental Vitamin D and Calcium on Oxidative DNA Damage Marker in Normal Colorectal Mucosa: A Randomized Clinical Trial. Cancer Epidemiol. Biomark. Prev. Publ. Am. Assoc. Cancer Res. Cosponsored Am. Soc. Prev. Oncol. 2010, 19, 280–291.
- Peehl, D.M.; Shinghal, R.; Nonn, L.; Seto, E.; Krishnan, A.V.; Brooks, J.D.; Feldman, D. Molecular Activity of 1,25-Dihydroxyvitamin D3 in Primary Cultures of Human Prostatic Epithelial Cells Revealed by CDNA Microarray Analysis. J. Steroid Biochem. Mol. Biol. 2004, 92, 131–141.
- Lambert, J.R.; Kelly, J.A.; Shim, M.; Huffer, W.E.; Nordeen, S.K.; Baek, S.J.; Eling, T.E.; Lucia, M.S. Prostate Derived Factor in Human Prostate Cancer Cells: Gene Induction by Vitamin D via a P53-Dependent Mechanism and Inhibition of Prostate Cancer Cell Growth. J. Cell. Physiol. 2006, 208, 566–574.
- Swami, S.; Raghavachari, N.; Muller, U.R.; Bao, Y.P.; Feldman, D. Vitamin D Growth Inhibition of Breast Cancer Cells: Gene Expression Patterns Assessed by CDNA Microarray. Breast Cancer Res. Treat. 2003, 80, 49–62.
- Bao, B.-Y.; Ting, H.-J.; Hsu, J.-W.; Lee, Y.-F. Protective Role of 1α, 25-Dihydroxyvitamin D3 against Oxidative Stress in Nonmalignant Human Prostate Epithelial Cells. Int. J. Cancer 2008, 122, 2699–2706.
- Zhang, X.; Li, P.; Bao, J.; Nicosia, S.V.; Wang, H.; Enkemann, S.A.; Bai, W. Suppression of Death Receptor-Mediated Apoptosis by 1,25-Dihydroxyvitamin D3 Revealed by Microarray Analysis. J. Biol. Chem. 2005, 280, 35458–35468.
- Teixeira, T.M.; da Costa, D.C.; Resende, A.C.; Soulage, C.O.; Bezerra, F.F.; Daleprane, J.B. Activation of Nrf2-Antioxidant Signaling by 1,25-Dihydroxycholecalciferol Prevents Leptin-Induced Oxidative Stress and Inflammation in Human Endothelial Cells. J. Nutr. 2017, 147, 506–513.
- Nakai, K.; Fujii, H.; Kono, K.; Goto, S.; Kitazawa, R.; Kitazawa, S.; Hirata, M.; Shinohara, M.; Fukagawa, M.; Nishi, S. Vitamin D Activates the Nrf2-Keap1 Antioxidant Pathway and Ameliorates Nephropathy in Diabetic Rats. Am. J. Hypertens. 2014, 27, 586–595.
- Kim, H.K.; Andreazza, A.C.; Yeung, P.Y.; Isaacs-Trepanier, C.; Young, L.T. Oxidation and Nitration in Dopaminergic Areas of the Prefrontal Cortex from Patients with Bipolar Disorder and Schizophrenia. J. Psychiatry Neurosci. JPN 2014, 39, 276–285.
- Scaini, G.; Rezin, G.T.; Carvalho, A.F.; Streck, E.L.; Berk, M.; Quevedo, J. Mitochondrial Dysfunction in Bipolar Disorder: Evidence, Pathophysiology and Translational Implications. Neurosci. Biobehav. Rev. 2016, 68, 694–713.
- Silvagno, F.; Consiglio, M.; Foglizzo, V.; Destefanis, M.; Pescarmona, G. Mitochondrial Translocation of Vitamin D Receptor Is Mediated by the Permeability Transition Pore in Human Keratinocyte Cell Line. PLoS ONE 2013, 8, e54716.
- Silvagno, F.; De Vivo, E.; Attanasio, A.; Gallo, V.; Mazzucco, G.; Pescarmona, G. Mitochondrial Localization of Vitamin D Receptor in Human Platelets and Differentiated Megakaryocytes. PLoS ONE 2010, 5, e8670.
- Berridge, M.J. Vitamin D Deficiency and Diabetes. Biochem. J. 2017, 474, 1321–1332.
- Qatanani, M.; Lazar, M.A. Mechanisms of Obesity-Associated Insulin Resistance: Many Choices on the Menu. Genes Dev. 2007, 21, 1443–1455.
- Verdile, G.; Keane, K.N.; Cruzat, V.F.; Medic, S.; Sabale, M.; Rowles, J.; Wijesekara, N.; Martins, R.N.; Fraser, P.E.; Newsholme, P. Inflammation and Oxidative Stress: The Molecular Connectivity between Insulin Resistance, Obesity, and Alzheimer’s Disease. Available online: (accessed on 17 January 2019).
- Rains, J.L.; Jain, S.K. OXIDATIVE STRESS, INSULIN SIGNALING AND DIABETES. Free Radic. Biol. Med. 2011, 50, 567–575.
- Ricciardi, C.J.; Bae, J.; Esposito, D.; Komarnytsky, S.; Hu, P.; Chen, J.; Zhao, L. 1,25-Dihydroxyvitamin D3/Vitamin D Receptor Suppresses Brown Adipocyte Differentiation and Mitochondrial Respiration. Eur. J. Nutr. 2015, 54, 1001–1012.
- Ricca, C.; Aillon, A.; Bergandi, L.; Alotto, D.; Castagnoli, C.; Silvagno, F. Vitamin D Receptor Is Necessary for Mitochondrial Function and Cell Health. Int. J. Mol. Sci. 2018, 19, 1672.
- Seril, D.N.; Liao, J.; Yang, G.-Y.; Yang, C.S. Oxidative Stress and Ulcerative Colitis-Associated Carcinogenesis: Studies in Humans and Animal Models. Carcinogenesis 2003, 24, 353–362.
- Blasiak, J.; Arabski, M.; Krupa, R.; Wozniak, K.; Zadrozny, M.; Kasznicki, J.; Zurawska, M.; Drzewoski, J. DNA Damage and Repair in Type 2 Diabetes Mellitus. Mutat. Res. 2004, 554, 297–304.
- Yoshino, M.; Yoshino, J.; Kayser, B.D.; Patti, G.; Franczyk, M.P.; Mills, K.F.; Sindelar, M.; Pietka, T.; Patterson, B.W.; Imai, S.-I.; et al. Nicotinamide Mononucleotide Increases Muscle Insulin Sensitivity in Prediabetic Women. Science 2021.
- Kumar, V.; Agrawal, R.; Pandey, A.; Kopf, S.; Hoeffgen, M.; Kaymak, S.; Bandapalli, O.R.; Gorbunova, V.; Seluanov, A.; Mall, M.A.; et al. Compromised DNA Repair Is Responsible for Diabetes-Associated Fibrosis. EMBO J. 2020, 39, e103477.
- Hishikawa, A.; Hayashi, K.; Yoshimoto, N.; Nakamichi, R.; Homma, K.; Itoh, H. DNA Damage and Expression of DNA Methylation Modulators in Urine-Derived Cells of Patients with Hypertension and Diabetes. Sci. Rep. 2020, 10, 3377.
- Hishikawa, A.; Hayashi, K.; Abe, T.; Kaneko, M.; Yokoi, H.; Azegami, T.; Nakamura, M.; Yoshimoto, N.; Kanda, T.; Itoh, H.; et al. Decreased KAT5 Expression Impairs DNA Repair and Induces Altered DNA Methylation in Kidney Podocytes. Cell Rep. 2019, 26.
- Lin, J.; Song, T.; Li, C.; Mao, W. GSK-3β in DNA Repair, Apoptosis, and Resistance of Chemotherapy, Radiotherapy of Cancer. Biochim. Biophys. Acta Mol. Cell Res. 2020, 1867, 118659.
- Jope, R.S.; Johnson, G.V.W. The Glamour and Gloom of Glycogen Synthase Kinase-3. Trends Biochem. Sci. 2004, 29, 95–102.
- Iside, C.; Scafuro, M.; Nebbioso, A.; Altucci, L. SIRT1 Activation by Natural Phytochemicals: An Overview. Front. Pharmacol. 2020, 11, 1225.
- Nebbioso, M.; Lambiase, A.; Armentano, M.; Tucciarone, G.; Sacchetti, M.; Greco, A.; Alisi, L. Diabetic Retinopathy, Oxidative Stress, and Sirtuins: An in Depth Look in Enzymatic Patterns and New Therapeutic Horizons. Surv. Ophthalmol. 2021.
- Alves-Fernandes, D.K.; Jasiulionis, M.G. The Role of SIRT1 on DNA Damage Response and Epigenetic Alterations in Cancer. Int. J. Mol. Sci. 2019, 20, 3153.
- Merkel, P.; Khoury, N.; Bertolotto, C.; Perfetti, R. Insulin and Glucose Regulate the Expression of the DNA Repair Enzyme XPD. Mol. Cell. Endocrinol. 2003, 201, 75–85.
- Ting, H.-J.; Yasmin-Karim, S.; Yan, S.-J.; Hsu, J.-W.; Lin, T.-H.; Zeng, W.; Messing, J.; Sheu, T.-J.; Bao, B.-Y.; Li, W.X.; et al. A Positive Feedback Signaling Loop between ATM and the Vitamin D Receptor Is Critical for Cancer Chemoprevention by Vitamin D. Cancer Res. 2012, 72, 958–968.
- Akhter, J.; Chen, X.; Bowrey, P.; Bolton, E.J.; Morris, D.L. Vitamin D3 Analog, EB1089, Inhibits Growth of Subcutaneous Xenografts of the Human Colon Cancer Cell Line, LoVo, in a Nude Mouse Model. Dis. Colon Rectum 1997, 40, 317–321.
- Jiang, F.; Li, P.; Fornace, A.J.; Nicosia, S.V.; Bai, W. G2/M Arrest by 1,25-Dihydroxyvitamin D3 in Ovarian Cancer Cells Mediated through the Induction of GADD45 via an Exonic Enhancer. J. Biol. Chem. 2003, 278, 48030–48040.
- Gonzalo, S. Novel Roles of 1α,25(OH)2D3 on DNA Repair Provide New Strategies for Breast Cancer Treatment. J. Steroid Biochem. Mol. Biol. 2014, 144PA, 59–64.
- Samuel, S.; Sitrin, M.D. Vitamin D’s Role in Cell Proliferation and Differentiation. Nutr. Rev. 2008, 66, S116–S124.
- Larriba, M.J.; González-Sancho, J.M.; Barbáchano, A.; Niell, N.; Ferrer-Mayorga, G.; Muñoz, A. Vitamin D Is a Multilevel Repressor of Wnt/β-Catenin Signaling in Cancer Cells. Cancers 2013, 5, 1242–1260.
- Pendás-Franco, N.; García, J.M.; Peña, C.; Valle, N.; Pálmer, H.G.; Heinäniemi, M.; Carlberg, C.; Jiménez, B.; Bonilla, F.; Muñoz, A.; et al. DICKKOPF-4 Is Induced by TCF/Beta-Catenin and Upregulated in Human Colon Cancer, Promotes Tumour Cell Invasion and Angiogenesis and Is Repressed by 1alpha,25-Dihydroxyvitamin D3. Oncogene 2008, 27, 4467–4477.
- An, B.-S.; Tavera-Mendoza, L.E.; Dimitrov, V.; Wang, X.; Calderon, M.R.; Wang, H.-J.; White, J.H. Stimulation of Sirt1-Regulated FoxO Protein Function by the Ligand-Bound Vitamin D Receptor. Mol. Cell. Biol. 2010, 30, 4890–4900.
- Yang, L.; Yang, J.; Venkateswarlu, S.; Ko, T.; Brattain, M.G. Autocrine TGFbeta Signaling Mediates Vitamin D3 Analog-Induced Growth Inhibition in Breast Cells. J. Cell. Physiol. 2001, 188, 383–393.
- Chen, A.; Davis, B.H.; Sitrin, M.D.; Brasitus, T.A.; Bissonnette, M. Transforming Growth Factor-Beta 1 Signaling Contributes to Caco-2 Cell Growth Inhibition Induced by 1,25(OH)(2)D(3). Am. J. Physiol. Gastrointest. Liver Physiol. 2002, 283, G864–G874.
- de Caestecker, M.P.; Piek, E.; Roberts, A.B. Role of Transforming Growth Factor-Beta Signaling in Cancer. J. Natl. Cancer Inst. 2000, 92, 1388–1402.
- Buschke, S.; Stark, H.-J.; Cerezo, A.; Prätzel-Wunder, S.; Boehnke, K.; Kollar, J.; Langbein, L.; Heldin, C.-H.; Boukamp, P. A Decisive Function of Transforming Growth Factor-β/Smad Signaling in Tissue Morphogenesis and Differentiation of Human HaCaT Keratinocytes. Mol. Biol. Cell 2011, 22, 782–794.
- Ding, Z.; Wu, C.-J.; Chu, G.C.; Xiao, Y.; Ho, D.; Zhang, J.; Perry, S.R.; Labrot, E.S.; Wu, X.; Lis, R.; et al. SMAD4-Dependent Barrier Constrains Prostate Cancer Growth and Metastatic Progression. Nature 2011, 470, 269–273.
- Lee, H.J.; Liu, H.; Goodman, C.; Ji, Y.; Maehr, H.; Uskokovic, M.; Notterman, D.; Reiss, M.; Suh, N. Gene Expression Profiling Changes Induced by a Novel Gemini Vitamin D Derivative during the Progression of Breast Cancer. Biochem. Pharmacol. 2006, 72, 332–343.
- Krishnan, A.V.; Shinghal, R.; Raghavachari, N.; Brooks, J.D.; Peehl, D.M.; Feldman, D. Analysis of Vitamin D-Regulated Gene Expression in LNCaP Human Prostate Cancer Cells Using CDNA Microarrays. The Prostate 2004, 59, 243–251.
- Lin, R.; Nagai, Y.; Sladek, R.; Bastien, Y.; Ho, J.; Petrecca, K.; Sotiropoulou, G.; Diamandis, E.P.; Hudson, T.J.; White, J.H. Expression Profiling in Squamous Carcinoma Cells Reveals Pleiotropic Effects of Vitamin D3 Analog EB1089 Signaling on Cell Proliferation, Differentiation, and Immune System Regulation. Mol. Endocrinol. 2002, 16, 1243–1256.
- Pálmer, H.G.; Sánchez-Carbayo, M.; Ordóñez-Morán, P.; Larriba, M.J.; Cordón-Cardó, C.; Muñoz, A. Genetic Signatures of Differentiation Induced by 1alpha,25-Dihydroxyvitamin D3 in Human Colon Cancer Cells. Cancer Res. 2003, 63, 7799–7806.
- Kodach, L.L.; Wiercinska, E.; de Miranda, N.F.C.C.; Bleuming, S.A.; Musler, A.R.; Peppelenbosch, M.P.; Dekker, E.; van den Brink, G.R.; van Noesel, C.J.M.; Morreau, H.; et al. The Bone Morphogenetic Protein Pathway Is Inactivated in the Majority of Sporadic Colorectal Cancers. Gastroenterology 2008, 134, 1332–1341.
- Denduluri, S.K.; Idowu, O.; Wang, Z.; Liao, Z.; Yan, Z.; Mohammed, M.K.; Ye, J.; Wei, Q.; Wang, J.; Zhao, L.; et al. Insulin-like Growth Factor (IGF) Signaling in Tumorigenesis and the Development of Cancer Drug Resistance. Genes Dis. 2014, 2, 13–25.
- Colston, K.W.; Perks, C.M.; Xie, S.P.; Holly, J.M. Growth Inhibition of Both MCF-7 and Hs578T Human Breast Cancer Cell Lines by Vitamin D Analogues Is Associated with Increased Expression of Insulin-like Growth Factor Binding Protein-3. J. Mol. Endocrinol. 1998, 20, 157–162.
- Huynh, H.; Pollak, M.; Zhang, J.C. Regulation of Insulin-like Growth Factor (IGF) II and IGF Binding Protein 3 Autocrine Loop in Human PC-3 Prostate Cancer Cells by Vitamin D Metabolite 1,25(OH)2D3 and Its Analog EB1089. Int. J. Oncol. 1998, 13, 137–143.
- Sprenger, C.C.; Peterson, A.; Lance, R.; Ware, J.L.; Drivdahl, R.H.; Plymate, S.R. Regulation of Proliferation of Prostate Epithelial Cells by 1,25-Dihydroxyvitamin D3 Is Accompanied by an Increase in Insulin-like Growth Factor Binding Protein-3. J. Endocrinol. 2001, 170, 609–618.
- Kovalenko, P.L.; Zhang, Z.; Cui, M.; Clinton, S.K.; Fleet, J.C. 1,25 Dihydroxyvitamin D-Mediated Orchestration of Anticancer, Transcript-Level Effects in the Immortalized, Non-Transformed Prostate Epithelial Cell Line, RWPE1. BMC Genom. 2010, 11, 26.
- Boyle, B.J.; Zhao, X.Y.; Cohen, P.; Feldman, D. Insulin-like Growth Factor Binding Protein-3 Mediates 1 Alpha,25-Dihydroxyvitamin d(3) Growth Inhibition in the LNCaP Prostate Cancer Cell Line through P21/WAF1. J. Urol. 2001, 165, 1319–1324.
- Nickerson, T.; Huynh, H. Vitamin D Analogue EB1089-Induced Prostate Regression Is Associated with Increased Gene Expression of Insulin-like Growth Factor Binding Proteins. J. Endocrinol. 1999, 160, 223–229.
- Zhang, Y.; Wang, X. Targeting the Wnt/β-Catenin Signaling Pathway in Cancer. J. Hematol. Oncol. J. Hematol. Oncol. 2020, 13, 165.
- Macleod, K. Tumor Suppressor Genes. Curr. Opin. Genet. Dev. 2000, 10, 81–93.
- Schneikert, J.; Behrens, J. The Canonical Wnt Signalling Pathway and Its APC Partner in Colon Cancer Development. Gut 2007, 56, 417–425.
- Pálmer, H.G.; González-Sancho, J.M.; Espada, J.; Berciano, M.T.; Puig, I.; Baulida, J.; Quintanilla, M.; Cano, A.; de Herreros, A.G.; Lafarga, M.; et al. Vitamin D(3) Promotes the Differentiation of Colon Carcinoma Cells by the Induction of E-Cadherin and the Inhibition of Beta-Catenin Signaling. J. Cell Biol. 2001, 154, 369–387.
- Egan, J.B.; Thompson, P.A.; Vitanov, M.V.; Bartik, L.; Jacobs, E.T.; Haussler, M.R.; Gerner, E.W.; Jurutka, P.W. Vitamin D Receptor Ligands, Adenomatous Polyposis Coli, and the Vitamin D Receptor FokI Polymorphism Collectively Modulate Beta-Catenin Activity in Colon Cancer Cells. Mol. Carcinog. 2010, 49, 337–352.
- Xu, H.; Posner, G.H.; Stevenson, M.; Campbell, F.C. Apc(MIN) Modulation of Vitamin D Secosteroid Growth Control. Carcinogenesis 2010, 31, 1434–1441.
- Shah, S.; Islam, M.N.; Dakshanamurthy, S.; Rizvi, I.; Rao, M.; Herrell, R.; Zinser, G.; Valrance, M.; Aranda, A.; Moras, D.; et al. The Molecular Basis of Vitamin D Receptor and Beta-Catenin Crossregulation. Mol. Cell 2006, 21, 799–809.
- Aguilera, O.; Peña, C.; García, J.M.; Larriba, M.J.; Ordóñez-Morán, P.; Navarro, D.; Barbáchano, A.; López de Silanes, I.; Ballestar, E.; Fraga, M.F.; et al. The Wnt Antagonist DICKKOPF-1 Gene Is Induced by 1alpha,25-Dihydroxyvitamin D3 Associated to the Differentiation of Human Colon Cancer Cells. Carcinogenesis 2007, 28, 1877–1884.
- Ribatti, D.; Tamma, R.; Annese, T. Epithelial-Mesenchymal Transition in Cancer: A Historical Overview. Transl. Oncol. 2020, 13.
- Thiery, J.P.; Acloque, H.; Huang, R.Y.J.; Nieto, M.A. Epithelial-Mesenchymal Transitions in Development and Disease. Cell 2009, 139, 871–890.
- Hou, Y.-F.; Gao, S.-H.; Wang, P.; Zhang, H.-M.; Liu, L.-Z.; Ye, M.-X.; Zhou, G.-M.; Zhang, Z.-L.; Li, B.-Y. 1α,25(OH)₂D₃ Suppresses the Migration of Ovarian Cancer SKOV-3 Cells through the Inhibition of Epithelial-Mesenchymal Transition. Int. J. Mol. Sci. 2016, 17, 1285.
- Lungchukiet, P.; Sun, Y.; Kasiappan, R.; Quarni, W.; Nicosia, S.V.; Zhang, X.; Bai, W. Suppression of Epithelial Ovarian Cancer Invasion into the Omentum by 1α,25-Dihydroxyvitamin D3 and Its Receptor. J. Steroid Biochem. Mol. Biol. 2015, 148, 138–147.
- Liu, L.; Hu, Z.; Zhang, H.; Hou, Y.; Zhang, Z.; Zhou, G.; Li, B. Vitamin D Postpones the Progression of Epithelial Ovarian Cancer Induced by 7, 12-Dimethylbenz [a] Anthracene Both in Vitro and in Vivo. OncoTargets Ther. 2016, 9, 2365–2375.
- Abdelbaset-Ismail, A.; Pedziwiatr, D.; Suszyńska, E.; Sluczanowska-Glabowska, S.; Schneider, G.; Kakar, S.S.; Ratajczak, M.Z. Vitamin D3 Stimulates Embryonic Stem Cells but Inhibits Migration and Growth of Ovarian Cancer and Teratocarcinoma Cell Lines. J. Ovarian Res. 2016, 9, 26.
- Chen, Y.; Sun, Z.; Xu, J.; Wang, P.; Tang, J.; Shi, X.; Liu, J.; Ren, F.; Xu, L. Vitamin D and DDX4 Regulate the Proliferation and Invasion of Ovarian Cancer Cells. Oncol. Lett. 2018, 16, 905–909.
- Kriebitzsch, C.; Verlinden, L.; Eelen, G.; Tan, B.K.; Van Camp, M.; Bouillon, R.; Verstuyf, A. The Impact of 1,25(OH)2D3 and Its Structural Analogs on Gene Expression in Cancer Cells—A Microarray Approach. Anticancer Res. 2009, 29, 3471–3483.
- Díaz, G.D.; Paraskeva, C.; Thomas, M.G.; Binderup, L.; Hague, A. Apoptosis Is Induced by the Active Metabolite of Vitamin D3 and Its Analogue EB1089 in Colorectal Adenoma and Carcinoma Cells: Possible Implications for Prevention and Therapy. Cancer Res. 2000, 60, 2304–2312.
- Pan, L.; Matloob, A.F.; Du, J.; Pan, H.; Dong, Z.; Zhao, J.; Feng, Y.; Zhong, Y.; Huang, B.; Lu, J. Vitamin D Stimulates Apoptosis in Gastric Cancer Cells in Synergy with Trichostatin A/Sodium Butyrate-Induced and 5-Aza-2′-Deoxycytidine-Induced PTEN Upregulation. FEBS J. 2010, 277, 989–999.
- Sergeev, I.N. Vitamin D and Cellular Ca2+ Signaling in Breast Cancer. Anticancer Res. 2012, 32, 299–302.
- Sharma, K.; Goehe, R.W.; Di, X.; Hicks, M.A.; Torti, S.V.; Torti, F.M.; Harada, H.; Gewirtz, D.A. A Novel Cytostatic Form of Autophagy in Sensitization of Non-Small Cell Lung Cancer Cells to Radiation by Vitamin D and the Vitamin D Analog, EB 1089. Autophagy 2015, 10, 2346–2361.
- Wilson, E.N.; Bristol, M.L.; Di, X.; Maltese, W.A.; Koterba, K.; Beckman, M.J.; Gewirtz, D.A. A Switch between Cytoprotective and Cytotoxic Autophagy in the Radio Sensitization of Breast Tumor Cells by Chloroquine and Vitamin D. Horm. Cancer 2011, 2, 272–285.
- Høyer-Hansen, M.; Bastholm, L.; Mathiasen, I.S.; Elling, F.; Jäättelä, M. Vitamin D Analog EB1089 Triggers Dramatic Lysosomal Changes and Beclin 1-Mediated Autophagic Cell Death. Cell Death Differ. 2005, 12, 1297–1309.
- Lisse, T.S.; Liu, T.; Irmler, M.; Beckers, J.; Chen, H.; Adams, J.S.; Hewison, M. Gene Targeting by the Vitamin D Response Element Binding Protein Reveals a Role for Vitamin D in Osteoblast MTOR Signaling. FASEB J. 2011, 25, 937–947.
- Lisse, T.S.; Hewison, M. Vitamin D. Cell Cycle 2011, 10, 1888–1889.
- Rajabi, M.; Mousa, S.A. The Role of Angiogenesis in Cancer Treatment. Biomedicines 2017, 5, 34.
- Ben-Shoshan, M.; Amir, S.; Dang, D.T.; Dang, L.H.; Weisman, Y.; Mabjeesh, N.J. 1alpha,25-Dihydroxyvitamin D3 (Calcitriol) Inhibits Hypoxia-Inducible Factor-1/Vascular Endothelial Growth Factor Pathway in Human Cancer Cells. Mol. Cancer Ther. 2007, 6, 1433–1439.
- Chung, I.; Han, G.; Seshadri, M.; Gillard, B.M.; Yu, W.; Foster, B.A.; Trump, D.L.; Johnson, C.S. Role of Vitamin D Receptor in the Antiproliferative Effects of Calcitriol in Tumor-Derived Endothelial Cells and Tumor Angiogenesis in Vivo. Cancer Res. 2009, 69, 967–975.
- Wong, G.; Lim, W.H.; Lewis, J.; Craig, J.C.; Turner, R.; Zhu, K.; Lim, E.M.; Prince, R. Vitamin D and Cancer Mortality in Elderly Women. BMC Cancer 2015, 15, 106.


