 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Elżbieta Poniedziałek-Czajkowska | + 3736 word(s) | 3736 | 2021-06-28 08:26:35 | | | |
| 2 | Vivi Li | + 446 word(s) | 4182 | 2021-07-16 06:04:23 | | |
Video Upload Options
The possibility of prophylaxis of hypertensive disorders of pregnancy (HDPs) such as preeclampsia (PE) and pregnancy-induced hypertension (PIH) is of interest due to the unpredictable course of these diseases and the risks they carry for both mother and fetus. It has been proven that their development is associated with the presence of the placenta, and the processes that initiate it begin at the time of the abnormal invasion of the trophoblast in early pregnancy. The ideal HDPs prophylaxis should alleviate the influence of risk factors and, at the same time, promote physiological trophoblast invasion and maintain the physiologic endothelium function without any harm to both mother and fetus. So far, aspirin is the only effective and recommended pharmacological agent for the prevention of HDPs in high-risk groups. Metformin is a hypoglycemic drug with a proven protective effect on the cardiovascular system. Respecting the anti-inflammatory properties of metformin and its favorable impact on the endothelium, it seems to be an interesting option for HDPs prophylaxis.
1. Metformin
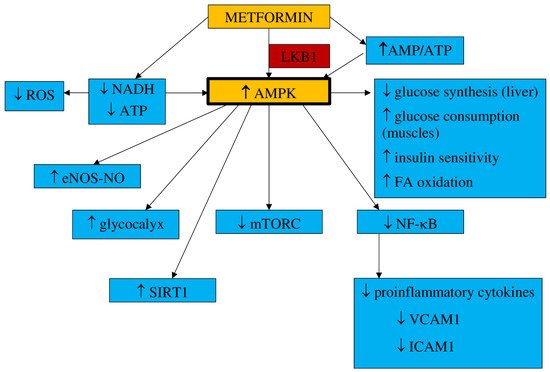
1.1. Pharmacokinetics and Mechanism of Action
- a.
-
Inflammation and oxidative stress
- b.
-
NO synthesis
- c.
-
Endothelial senescence and apoptosis
- d.
-
Vascular integrity
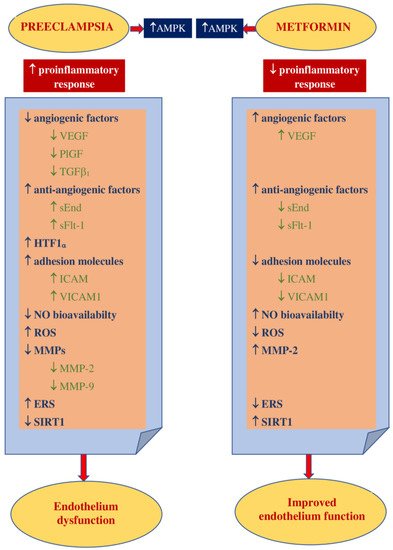
1.2. Impact on Preeclampsia Pathophysiology
2. Metformin in Preventing Hypertensive Disorders of Pregnancy
| Studied Group | Size of Groups | Metformin Dose | GA at Entry to the Study | PIH and PE | Authors |
|---|---|---|---|---|---|
| GDM high risk | SG: metformin—24 CG:no treatment—25 |
500–1000 mg | 14th week | PE SG: 0% (0) CG: 8.7% (2) p = 0.049 |
Brink et al., 2018 [67] |
| GDM | SG: metformin—43 CG:insulin alone—57 |
500–2500 mg | 20th–36th week | PIH SG: 18.6% (8) CG: 24% (18) NS PE SG: 0% (0) CG: 8% (6) p = 0.05 |
Ainuddin et al., 2014 [68] |
| GDM | SG: metformin 110 CG insulin 107 |
500–2000 mg | 22nd–34th week | PIH SG: 1.8% (2) CG: 3.7% (4) p = 0.42, RR 0.5 95% CI 0.1–2.7 PE SG: 4.6% (5) CG: 9.4% (10) p = 0.19, RR 0.5 95% CI 0.2–1.4 |
Tertti et al., 2013 [69] |
| GDM | SG metformin: 86 CG insulin: 80 |
1000–2500 mg | 20th–34th week | PIH SG: 5% (4) CG 13.8% (11) p = 0.058, RR 0.4 95% CI 0.1–1.1 PE SG: 6.3% (5) CG: 8.8% (7) p = 0.548, RR 0.7 95% CI 0.2–2.22 |
Niromanesh et al., 2012 [70] |
| DMt.2 | SG: metformin—233 CG: insulin—240 |
2000 mg | 6th–22th week | PIH SG: 5% (13) CG 6% (15) p = 0.82, RR 0.92 95% CI 0.46–1.8. PE SG: 15% (37) CG 12% (30) p = 0.29, RR 1.27 95% CI 0.82–1.92 Chronic HT ST: 8% (20) CG: 9% (22) p = 0.68, RR 0.89 95% CI 0.51–1.56 |
Feig et al., 2020 [71] |
| DMt.2 | SG: metformin alone—16 CG: insulin alone 100 |
500–2500 mg | about 10th week | PIH SG: 6.2% (1) CG: 36% (36) p= 0.020 PE SG: 25% (4) CG: 17% (17) p = 0.084 |
Ainuddin et al., 2015 [72] |
| Obesity | SG: metformin—171 CG: placebo—186 |
1000 mg | <20th week | PE SG: 3.5% (6) CG: 4.8% (9) p = 0.01, RR 0.17 95% CI 0.10–1.41 |
Nascimento et al., 2020 [73] |
| Obesity (35 kg/m2) | SG: metformin—202 CG: placebo—198 |
1000–3000 mg | 12th–18th week | PIH SG: 6.4% (13) CG: 6.7% (13) p = 0.93, RR 0.96 95% CI 0.43–2.13 PE SG: 3% (6) CG: 11.3% (13) p = 0.001, RR 0.24 95% CI 0.10–0.61 |
Syngelaki et al., 2016 [74] |
| Obesity (BMI > 30 kg/m2) | SG: metformin—221 CG: placebo—222 |
500–2500 mg | 12th–16th week | PIH SG: 10% (21) CG 6% (14) p= 0.22, RR 1.56 95% CI 0.77–3.15. PE SG: 3% (7) CG 1% (3) p = 0.21, RR 2.39 95% 0.61–9.36 |
Chiswick et al., 2015 [42] |
| PCOS | SG: metformin—238 CG: placebo—240 |
1000–2000 mg | in the 1st trimester | PE SG: 3% (8) CG: 7% (17) p = 0.10, RR 0.46 95% CI 0.17–1.15 |
Løvvik et al., 2019 [75] |
| Studied Group | Comparison | Number of Participants | Metformin Impact on PIH/PE | Authors |
|---|---|---|---|---|
| GDM | metformin vs. insulin | 1260 | PIH RR 0.56 95% CI 0.37–0.85 PE RR 0.83 95% CI 0.60–1.14 PE RR 0.74 95% CI 0.09–6.28 |
Kalafat et al., 2018 [80] |
| Obesity | metformin vs. placebo | 840 | ||
| GDM | Metformin vs. insulin | 2165 | ↓PIH RR 0.56 95% CI 0.37–0.85 |
Butalia et al., 2017 [81] |
| GDM | metformin vs. insulin | 1556 | ↓HDPs RR 0.82 95% CI 0.67–1.0 |
Feng et al., 2017 [82] |
| GDM | metformin vs. insulin | 1110 | ↓PIH RR 0.53 95% CI 0.31–0.90 PE RR 0.81 95% CI 0.55–1.17, |
Li et al., 2015 [83] |
| 1634 | ||||
| GDM | metformin vs. insulin | 1110 | ↓PIH RR 0.55 95% CI 0.31–0.91 PE RR 0.84 95% CI 0.57–1.23 |
Poolsup et al., 2014 [84] |
| 1299 | ||||
| GDM | metformin vs. insulin | 1712 | PE RR = 0.82 95% CI 0.56–1.2 |
Zhu et al., 2014 [85] |
| GDM | metformin vs. insulin | 1110 | ↓PIH RR 0.52 95%CI 0.30–0.90 |
Gui et al., 2013 [86] |
| Obesity | metformin vs. no-treatment | 840 614 308 |
PIH (obesity) RR 1.24 95% CI 0.76–2.02 ↓PE (obesity) RR 0.51 95% CI 0.26–0.98 ↓PIH (PCOS) RR 0.37 95% CI 0.25–0.57 PE (PCOS) RR 1.96 95% CI 0.81–4.77 ↓PIH (GDM) RR 0.53 95% CI 0.31–0.90 PE (GDM) RR 0.70 95% CI 0.45–1.10 |
Nascimento et al., 2018 [79] |
| PCOS | ||||
| GDM | metformin vs. insulin | 1120 1120 |
||
| Obesity | metformin vs. no-treatment or placebo | 1034 | PIH RR 1.02 95% CI 0.54–1.94 PE RR 0.74 95% CI 0.09–6.28 |
Dodd et al., 2018 [87] |
| PCOS | metformin vs. no-treatment or placebo | 929 | PE RR 0.92 95% CI 0.28–3.00 |
Feng et al., 2015 [88] |
| PCOS | metformin vs. no-treatment or placebo | 878 | ↓PE RR 0.53 95% CI 0.30–0.95 |
Zheng et al., 2013 [89] |
References
- Rowan, J.A.; Hague, W.M.; Gao, W.; Battin, M.R.; Moore, M.P. MiG Trial Investigators. Metformin versus insulin for the treatment of gestational diabetes. N. Engl. J. Med. 2008, 358, 2003–2015.
- Graham, G.G.; Punt, J.; Arora, M.; Day, R.O.; Doogue, M.P.; Duong, J.K.; Furlong, T.J.; Greenfield, J.R.; Greenup, L.C.; Kirkpatrick, C.M.; et al. Clinical pharmacokinetics of metformin. Clin. Pharmacokinet. 2011, 50, 81–98.
- Foretz, M.; Guigas, B.; Bertrand, L.; Pollak, M.; Viollet, B. Metformin: From mechanisms of action to therapies. Cell Metab. 2014, 20, 953–966.
- He, L.; Wondisford, F.E. Metformin action: Concentrations matter. Cell Metab. 2015, 21, 159–162.
- El-Mir, M.Y.; Nogueira, V.; Fontaine, E.; Avéret, N.; Rigoulet, M.; Leverve, X. Dimethylbiguanide inhibits cell respiration via an indirect effect targeted on the respiratory chain complex I. J. Biol. Chem. 2000, 275, 223–228.
- Oliveras-Ferraros, C.; Vazquez-Martin, A.; Menendez, J.A. Genome-wide inhibitory impact of the AMPK activator metformin on [kinesins, tubulins, histones, auroras and polo-like kinases] M-phase cell cycle genes in human breast cancer cells. Cell Cycle 2009, 8, 1633–1636.
- Zhou, G.; Myers, R.; Li, Y.; Chen, Y.; Shen, X.; Fenyk-Melody, J.; Wu, M.; Ventre, J.; Doebber, T.; Fujii, N.; et al. Role of AMP-activated protein kinase in mechanism of metformin action. J. Clin. Investig. 2001, 108, 1167–1174.
- Shaw, R.J.; Lamia, K.A.; Vasquez, D.; Koo, S.H.; Bardeesy, N.; Depinho, R.A.; Montminy, M.; Cantley, L.C. The kinase LKB1 mediates glucose homeostasis in liver and therapeutic effects of metformin. Science 2005, 310, 1642–1646.
- Zou, M.H.; Kirkpatrick, S.S.; Davis, B.J.; Nelson, J.S.; Wiles, W.G., IV; Schlattner, U.; Neumann, D.; Brownlee, M.; Freeman, M.B.; Goldman, M.H. Activation of the AMP-activated protein kinase by the anti-diabetic drug metformin in vivo. Role of mitochondrial reactive nitrogen species. J. Biol. Chem. 2004, 279, 43940–43951.
- Foretz, M.; Hébrard, S.; Leclerc, J.; Zarrinpashneh, E.; Soty, M.; Mithieux, G.; Sakamoto, K.; Andreelli, F.; Viollet, B. Metformin inhibits hepatic gluconeogenesis in mice independently of the LKB1/AMPK pathway via a decrease in hepatic energy state. J. Clin. Investig. 2010, 120, 2355–2369.
- Madiraju, A.K.; Erion, D.M.; Rahimi, Y.; Zhang, X.M.; Braddock, D.T.; Albright, R.A.; Prigaro, B.J.; Wood, J.L.; Bhanot, S.; MacDonald, M.J.; et al. Metformin suppresses gluconeogenesis by inhibiting mitochondrial glycerophosphate dehydrogenase. Nature 2014, 510, 542–546.
- Luo, Z.; Zang, M.; Guo, W. AMPK as a metabolic tumor suppressor: Control of metabolism and cell growth. Future Oncol. 2010, 6, 457–470.
- Bost, F.; Sahra, I.B.; Le Marchand-Brustel, Y.; Tanti, J.F. Metformin and cancer therapy. Curr. Opin. Oncol. 2012, 24, 103–108.
- Dowling, R.J.; Zakikhani, M.; Fantus, I.G.; Pollak, M.; Sonenberg, N. Metformin inhibits mammalian target of rapamycin-dependent translation initiation in breast cancer cells. Cancer Res. 2007, 67, 10804108012.
- Ben Sahra, I.; Regazzetti, C.; Robert, G.; Laurent, K.; Le Marchand-Brustel, Y.; Auberger, P.; Tanti, J.F.; Giorgetti-Peraldi, S.; Bost, F. Metformin, independent of AMPK, induces mTOR inhibition and cell-cycle arrest through REDD1. Cancer Res. 2011, 71, 4366–4372.
- Scheen, A.J.; Esser, N.; Paquot, N. Antidiabetic agents: Potential anti-inflammatory activity beyond glucose control. Diabetes Metab. 2015, 41, 183–194.
- Saenz, A.; Fernandez-Esteban, I.; Mataix, A.; Ausejo, M.; Roque, M.; Moher, D. Metformin monotherapy for type 2 diabetes mellitus. Cochrane Database Syst. Rev. 2005, 3, CD002966.
- Abbasi, F.; Chu, J.W.; McLaughlin, T.; Lamendola, C.; Leary, E.T.; Reaven, G.M. Effect of metformin treatment on multiple cardiovascular disease risk factors in patients with type 2 diabetes mellitus. Metabolism 2004, 53, 159–164.
- De Aguiar, L.G.; Bahia, L.R.; Villela, N.; Laflor, C.; Sicuro, F.; Wiernsperger, N.; Bottino, D.; Bouskela, E. Metformin improves endothelial vascular reactivity in first-degree relatives of type 2 diabetic patients with metabolic syndrome and normal glucose tolerance. Diabetes Care 2006, 29, 1083–1089.
- Libby, P. Mechanisms of acute coronary syndromes and their implications for therapy. N. Engl. J. Med. 2013, 368, 2004–2013.
- Sena, C.M.; Matafome, P.; Louro, T.; Nunes, E.; Fernandes, R.; Seiça, R.M. Metformin restores endothelial function in aorta of diabetic rats. Br. J. Pharmacol. 2011, 163, 424–437.
- Li, S.N.; Wang, X.; Zeng, Q.T.; Feng, Y.B.; Cheng, X.; Mao, X.B.; Wang, T.H.; Deng, H.P. Metformin inhibits nuclear factor kappaB activation and decreases serum high-sensitivity C-reactive protein level in experimental atherogenesis of rabbits. Heart Vessels. 2009, 24, 446–453.
- Hattori, Y.; Suzuki, K.; Hattori, S.; Kasai, K. Metformin inhibits cytokine-induced nuclear factor kappaB activation via AMP-activated protein kinase activation in vascular endothelial cells. Hypertension 2006, 47, 1183–1888.
- Isoda, K.; Young, J.L.; Zirlik, A.; MacFarlane, L.A.; Tsuboi, N.; Gerdes, N.; Schönbeck, U.; Libby, P. Metformin inhibits proinflammatory responses and nuclear factor-kappaB in human vascular wall cells. Arterioscler. Thromb. Vasc. Biol. 2006, 26, 611–617.
- Hyun, B.; Shin, S.; Lee, A.; Lee, S.; Song, Y.; Ha, N.J.; Cho, K.H.; Kim, K. Metformin down-regulates TNF-α secretion via suppression of scavenger receptors in macrophages. Immune Netw. 2013, 13, 123–132.
- Gongol, B.; Marin, T.; Peng, I.C.; Woo, B.; Martin, M.; King, S.; Sun, W.; Johnson, D.A.; Chien, S.; Shyy, J.Y. AMPKα2 exerts its anti-inflammatory effects through PARP-1 and Bcl-6. Proc. Natl. Acad. Sci. USA 2013, 110, 3161–3166.
- Kim, S.A.; Choi, H.C. Metformin inhibits inflammatory response via AMPK-PTEN pathway in vascular smooth muscle cells. Biochem. Biophys. Res. Commun. 2012, 425, 866–872.
- Beisswenger, P.; Ruggiero-Lopez, D. Metformin inhibition of glycation processes. Diabetes Metab. 2003, 29, 6S95–6S103.
- Yan, S.F.; D’Agati, V.; Schmidt, A.M.; Ramasamy, R. Receptor for Advanced Glycation Endproducts (RAGE): A formidable force in the pathogenesis of the cardiovascular complications of diabetes & aging. Curr. Mol. Med. 2007, 7, 699–710.
- Di Marco, E.; Gray, S.P.; Jandeleit-Dahm, K. Diabetes alters activation and repression of pro- and anti-inflammatory signaling pathways in the vasculature. Front. Endocrinol. 2013, 4, 68.
- Mamputu, J.C.; Wiernsperger, N.F.; Renier, G. Antiatherogenic properties of metformin: The experimental evidence. Diabetes Metab. 2003, 29, 6S71–6S76.
- Saisho, Y. Metformin and inflammation: Its potential beyond glucose-lowering effect. Endocr. Metab. Immune Disord. Drug Targets 2015, 15, 196–205.
- Ouslimani, N.; Peynet, J.; Bonnefont-Rousselot, D.; Thérond, P.; Legrand, A.; Beaudeux, J.L. Metformin decreases intracellular production of reactive oxygen species in aortic endothelial cells. Metabolism 2005, 54, 829–834.
- Bakhashab, S.; Ahmed, F.W.; Schulten, H.J.; Bashir, A.; Karim, S.; Al-Malki, A.L.; Gari, M.A.; Abuzenadah, A.M.; Chaudhary, A.G.; Alqahtani, M.H.; et al. Metformin improves the angiogenic potential of human CD34⁺ cells co-incident with downregulating CXCL10 and TIMP1 gene expression and increasing VEGFA under hyperglycemia and hypoxia within a therapeutic window for myocardial infarction. Cardiovasc. Diabetol. 2016, 15, 27.
- Yu, J.W.; Deng, Y.P.; Han, X.; Ren, G.F.; Cai, J.; Jiang, G.J. Metformin improves the angiogenic functions of endothelial progenitor cells via activating AMPK/eNOS pathway in diabetic mice. Cardiovasc. Diabetol. 2016, 15, 88.
- Arunachalam, G.; Samuel, S.M.; Marei, I.; Ding, H.; Triggle, C.R. Metformin modulates hyperglycaemia-induced endothelial senescence and apoptosis through SIRT1. Br. J. Pharmacol. 2014, 171, 523–535.
- Kinaan, M.; Ding, H.; Triggle, C.R. Metformin: An old drug for the treatment of diabetes but a new drug for the protection of the endothelium. Med. Princ. Pract. 2015, 24, 401–415.
- Eskens, B.J.; Zuurbier, C.J.; van Haare, J.; Vink, H.; van Teeffelen, J.W. Effects of two weeks of metformin treatment on whole-body glycocalyx barrier properties in db/db mice. Cardiovasc. Diabetol. 2013, 12, 175.
- Vega, M.; Mauro, M.; Williams, Z. Direct toxicity of insulin on the human placenta and protection by metformin. Fertil. Steril. 2019, 111, 489–496.e5.
- Tenório, M.B.; Ferreira, R.C.; Moura, F.A.; Bueno, N.B.; de Oliveira, A.C.M.; Goulart, M.O.F. Cross-talk between oxidative stress and inflammation in preeclampsia. Oxid. Med. Cell Longev. 2019, 2019, 8238727.
- Han, C.S.; Herrin, M.A.; Pitruzzello, M.C.; Mulla, M.J.; Werner, E.F.; Pettker, C.M.; Flannery, C.A.; Abrahams, V.M. Glucose and metformin modulate human first trimester trophoblast function: A model and potential therapy for diabetes-associated uteroplacental insufficiency. Am. J. Reprod. Immunol. 2015, 73, 362–371.
- Chiswick, C.; Reynolds, R.M.; Denison, F.; Drake, A.J.; Forbes, S.; Newby, D.E.; Walker, B.R.; Quenby, S.; Wray, S.; Weeks, A.; et al. Effect of metformin on maternal and fetal outcomes in obese pregnant women (EMPOWaR): A randomised, double-blind, placebo-controlled trial. Lancet Diabetes Endocrinol. 2015, 3, 778–786.
- Hu, J.; Zhang, J.; Zhu, B. Protective effect of metformin on a rat model of lipopolysaccharide-induced preeclampsia. Fundam. Clin. Pharmacol. 2019, 33, 649–658.
- Shu, C.; Yan, D.; Chen, C.; Mo, Y.; Wu, L.; Gu, J.; Shah, N.K.; He, J.; Dong, S. Metformin exhibits its therapeutic effect in the treatment of pre-eclampsia via modulating the Met/H19/miR-148a-5p/P28 and Met/H19/miR-216-3p/EBI3 signaling pathways. Int. Immunopharmacol. 2019, 74, 105693.
- Correia-Branco, A.; Keating, E.; Martel, F. Involvement of mTOR, JNK and PI3K in the negative effect of ethanol and metformin on the human first-trimester extravillous trophoblast HTR-8/SVneo cell line. Eur. J. Pharmacol. 2018, 833, 16–24.
- Triggle, C.R.; Ding, H. Metformin is not just an antihyperglycaemic drug but also has protective effects on the vascular endothelium. Acta Physiol. 2017, 219, 138–151.
- Austgulen, R.; Lien, E.; Vince, G.; Redman, C.W. Increased maternal plasma levels of soluble adhesion molecules (ICAM-1, VCAM-1, E-selectin) in preeclampsia. Eur. J. Obstet. Gynecol. Reprod. Biol. 1997, 71, 53–58.
- Chaiworapongsa, T.; Romero, R.; Yoshimatsu, J.; Espinoza, J.; Kim, Y.M.; Park, K.; Kalache, K.; Edwin, S.; Bujold, E.; Gomez, R. Soluble adhesion molecule profile in normal pregnancy and pre-eclampsia. J. Matern. Fetal. Neonatal. Med. 2002, 12, 19–27.
- Brownfoot, F.C.; Hastie, R.; Hannan, N.J.; Cannon, P.; Tuohey, L.; Parry, L.J.; Senadheera, S.; Illanes, S.E.; Kaitu’u-Lino, T.J.; Tong, S. Metformin as a prevention and treatment for preeclampsia: Effects on soluble fms-like tyrosine kinase 1 and soluble endoglin secretion and endothelial dysfunction. Am. J. Obstet. Gynecol. 2016, 214, 356.e1–356.e15.
- Kaitu’u-Lino, T.J.; Brownfoot, F.C.; Beard, S.; Cannon, P.; Hastie, R.; Nguyen, T.V.; Binder, N.K.; Tong, S.; Hannan, N.J. Combining metformin and esomeprazole is additive in reducing sFlt-1 secretion and decreasing endothelial dysfunction-implications for treating preeclampsia. PLoS ONE 2018, 13, e0188845.
- Soobryan, N.; Murugesan, S.; Pandiyan, A.; Moodley, J.; Mackraj, I. Angiogenic dysregulation in pregnancy-related hypertension-A role for metformin. Reprod. Sci. 2018, 25, 1531–1539.
- Brownfoot, F.C.; Hastie, R.; Hannan, N.J.; Cannon, P.; Nguyen, T.V.; Tuohey, L.; Cluver, C.; Tong, S.; Kaitu’u-Lino, T.J. Combining metformin and sulfasalazine additively reduces the secretion of antiangiogenic factors from the placenta: Implications for the treatment of preeclampsia. Placenta 2020, 95, 78–83.
- Brouillet, S.; Hoffmann, P.; Feige, J.J.; Alfaidy, N. EG-VEGF: A key endocrine factor in placental development. Trends Endocrinol. Metab. 2012, 23, 501–508.
- Jena, M.K.; Sharma, N.R.; Petitt, M.; Maulik, D.; Nayak, N.R. Pathogenesis of preeclampsia and therapeutic approaches targeting the placenta. Biomolecules 2020, 10, 953.
- Wang, F.; Cao, G.; Yi, W.; Li, L.; Cao, X. Effect of metformin on a preeclampsia-like mouse model induced by high-fat diet. Biomed. Res. Int. 2019, 2019, 6547019.
- Sutton, E.F.; Gemmel, M.; Powers, R.W. Nitric oxide signaling in pregnancy and preeclampsia. Nitric. Oxide. 2020, 95, 55–62.
- Liao, Y.F.; Chen, L.L.; Zeng, T.S.; Li, Y.M.; Fan, Y.U.; Hu, L.J.; Ling, Y. Number of circulating endothelial progenitor cells as a marker of vascular endothelial function for type 2 diabetes. Vasc. Med. 2010, 15, 279–285.
- Asadian, S.; Alibabrdel, M.; Daei, N.; Cheraghi, H.; Maedeh Jafari, S.; Noshadirad, E.; Jabarpour, M.; Siavashi, V.; Nassiri, S.M. Improved angiogenic activity of endothelial progenitor cell in diabetic patients treated with insulin plus metformin. J. Cell Biochem. 2018.
- Ni, H.Z.; Liu, Z.; Sun, L.L.; Zhou, M.; Liu, C.; Li, W.D.; Li, X.Q. Metformin inhibits angiogenesis of endothelial progenitor cells via miR-221-mediated p27 expression and autophagy. Future Med. Chem. 2019, 11, 2263–2272.
- Li, W.D.; Li, N.P.; Song, D.D.; Rong, J.J.; Qian, A.M.; Li, X.Q. Metformin inhibits endothelial progenitor cell migration by decreasing matrix metalloproteinases, MMP-2 and MMP-9, via the AMPK/mTOR/autophagy pathway. Int. J. Mol. Med. 2017, 39, 1262–1268.
- Xu, C.; Bailly-Maitre, B.; Reed, J.C. Endoplasmic reticulum stress: Cell life and death decisions. J. Clin. Investig. 2005, 115, 2656–2664.
- Burton, G.J.; Yung, H.W.; Cindrova-Davies, T.; Charnock-Jones, D.S. Placental endoplasmic reticulum stress and oxidative stress in the pathophysiology of unexplained intrauterine growth restriction and early onset preeclampsia. Placenta 2009, 30, S43–S48.
- Burton, G.J.; Yung, H.W.; Murray, A.J. Mitochondrial-Endoplasmic reticulum interactions in the trophoblast: Stress and senescence. Placenta 2017, 52, 146–155.
- Suzuki, M.; Mizuuchi, M.; Baba, T.; Fujibe, Y.; Ino, Y.; Ishioka, S.; Saito, T. The roles of metformin and pravastatin on placental endoplasmic reticulum stress and placental growth factor in human villous-Like trophoblastic BeWo cells. Sapporo Med. J. 2018, 87, 75–84.
- Isaka, K.; Usuda, S.; Ito, H.; Sagawa, Y.; Nakamura, H.; Nishi, H.; Suzuki, Y.; Li, Y.F.; Takayama, M. Expression and activity of matrix metalloproteinase 2 and 9 in human trophoblasts. Placenta 2003, 24, 53–64.
- Rajendrasozhan, S.; Yang, S.R.; Kinnula, V.L.; Rahman, I. SIRT1, an antiinflammatory and antiaging protein, is decreased in lungs of patients with chronic obstructive pulmonary disease. Am. J. Respir. Crit. Care Med. 2008, 177, 861–870.
- Brink, H.S.; Alkemade, M.; van der Lely, A.J.; van der Linden, J. Metformin in women at high risk of gestational diabetes mellitus. Diabetes Metab. 2018, 44, 300–302.
- Ainuddin, J.; Karim, N.; Hasan, A.A.; Naqvi, S.A. Metformin versus insulin treatment in gestational diabetes in pregnancy in a developing country: A randomized control trial. Diabetes Res. Clin. Pract. 2015, 107, 290–299.
- Tertti, K.; Ekblad, U.; Koskinen, P.; Vahlberg, T.; Rönnemaa, T. Metformin vs. insulin in gestational diabetes. A randomized study characterizing metformin patients needing additional insulin. Diabetes Obes. Metab. 2013, 15, 246–251.
- Niromanesh, S.; Alavi, A.; Sharbaf, F.R.; Amjadi, N.; Moosavi, S.; Akbari, S. Metformin compared with insulin in the management of gestational diabetes mellitus: A randomized clinical trial. Diabetes Res. Clin. Pract. 2012, 98, 422–429.
- Feig, D.S.; Donovan, L.E.; Zinman, B.; Sanchez, J.J.; Asztalos, E.; Ryan, E.A.; Fantus, I.G.; Hutton, E.; Armson, A.B.; Lipscombe, L.L.; et al. Metformin in women with type 2 diabetes in pregnancy (MiTy): A multicentre, international, randomised, placebo-controlled trial. Lancet Diabetes Endocrinol. 2020, 8, 834–844.
- Ainuddin, J.A.; Karim, N.; Zaheer, S.; Ali, S.S.; Hasan, A.A. Metformin treatment in type 2 diabetes in pregnancy: An active controlled, parallel-group, randomized, open label study in patients with type 2 diabetes in pregnancy. J. Diabetes Res. 2015, 2015, 325851.
- Nascimento, I.B.D.; Sales, W.B.; Dienstmann, G.; Souza, M.L.R.; Fleig, R.; Silva, J.C. Metformin for prevention of cesarean delivery and large-for-gestational-age newborns in non-diabetic obese pregnant women: A randomized clinical trial. Arch. Endocrinol. Metab. 2020, 64, 290–297.
- Syngelaki, A.; Nicolaides, K.H.; Balani, J.; Hyer, S.; Akolekar, R.; Kotecha, R.; Pastides, A.; Shehata, H. Metformin versus placebo in obese pregnant women without diabetes mellitus. N. Engl. J. Med. 2016, 374, 434–443.
- Løvvik, T.S.; Carlsen, S.M.; Salvesen, Ø.; Steffensen, B.; Bixo, M.; Gómez-Real, F.; Lønnebotn, M.; Hestvold, K.V.; Zabielska, R.; Hirschberg, A.L.; et al. Use of metformin to treat pregnant women with polycystic ovary syndrome (PregMet2): A randomised, double-blind, placebo-controlled trial. Lancet Diabetes Endocrinol. 2019, 7, 256–266.
- Prejbisz, A.; Dobrowolski, P.; Kosiński, P.; Bomba-Opoń, D.; Adamczak, M.; Bekiesińska-Figatowska, M.; Kądziela, J.; Konopka, A.; Kostka-Jeziorny, K.; Kurnatowska, I.; et al. Management of hypertension in pregnancy: Prevention, diagnosis, treatment and long-term prognosis. Kardiol. Pol. 2019, 77, 757–806.
- Brown, M.A.; Magee, L.A.; Kenny, L.C.; Karumanchi, S.A.; McCarthy, F.P.; Saito, S.; Hall, D.R.; Warren, C.E.; Adoyi, G.; Ishaku, S.; et al. International Society for the Study of Hypertension in Pregnancy (ISSHP). The hypertensive disorders of pregnancy: ISSHP classification, diagnosis & management recommendations for international practice. Pregnancy Hypertens. 2018, 13, 291–310.
- Magee, L.A.; Pels, A.; Helewa, M.; Rey, E.; von Dadelszen, P.; SOGC Hypertension Guideline Committee. Diagnosis, evaluation, and management of the hypertensive disorders of pregnancy: Executive summary. J. Obstet. Gynaecol. Can. 2014, 36, 575–576.
- Nascimento, I.B.D.; Dienstmann, G.; de Souza, M.L.R.; Fleig, R.; Hoffmann, C.B.P.C.; Silva, J.C. Evaluation of preeclampsia results after use of metformin in gestation: Systematic review and meta-analysis. Rev. Bras. Ginecol. Obstet. 2018, 40, 713–721.
- Kalafat, E.; Sukur, Y.E.; Abdi, A.; Thilaganathan, B.; Khalil, A. Metformin for prevention of hypertensive disorders of pregnancy in women with gestational diabetes or obesity: Systematic review and meta-analysis of randomized trials. Ultrasound Obstet. Gynecol. 2018, 52, 706–714.
- Butalia, S.; Gutierrez, L.; Lodha, A.; Aitken, E.; Zakariasen, A.; Donovan, L. Short- and long-term outcomes of metformin compared with insulin alone in pregnancy: A systematic review and meta-analysis. Diabet. Med. 2017, 34, 27–36.
- Feng, Y.; Yang, H. Metformin-A potentially effective drug for gestational diabetes mellitus: A systematic review and meta-analysis. J. Matern. Fetal. Neonatal. Med. 2017, 30, 1874–1881.
- Li, G.; Zhao, S.; Cui, S.; Li, L.; Xu, Y.; Li, Y. Effect comparison of metformin with insulin treatment for gestational diabetes: A meta-analysis based on RCTs. Arch. Gynecol. Obstet. 2015, 292, 111–120.
- Poolsup, N.; Suksomboon, N.; Amin, M. Efficacy and safety of oral antidiabetic drugs in comparison to insulin in treating gestational diabetes mellitus: A meta-analysis. PLoS ONE 2014, 9, e109985.
- Zhu, B.; Zhang, L.; Fan, Y.Y.; Wang, L.; Li, X.G.; Liu, T.; Cao, Y.S.; Zhao, Z.G. Metformin versus insulin in gestational diabetes mellitus: A meta-analysis of randomized clinical trials. Ir. J. Med. Sci. 2016, 185, 371–381.
- Gui, J.; Liu, Q.; Feng, L. Metformin vs insulin in the management of gestational diabetes: A meta-analysis. PLoS ONE 2013, 8, e64585.
- Dodd, J.M.; Grivell, R.M.; Deussen, A.R.; Hague, W.M. Metformin for women who are overweight or obese during pregnancy for improving maternal and infant outcomes. Cochrane Database Syst. Rev. 2018, 7, CD010564.
- Feng, L.; Lin, X.F.; Wan, Z.H.; Hu, D.; Du, Y.K. Efficacy of metformin on pregnancy complications in women with polycystic ovary syndrome: A meta-analysis. Gynecol. Endocrinol. 2015, 31, 833–839.
- Zheng, J.; Shan, P.F.; Gu, W. The efficacy of metformin in pregnant women with polycystic ovary syndrome: A meta-analysis of clinical trials. J. Endocrinol. Investig. 2013, 36, 797–802.


