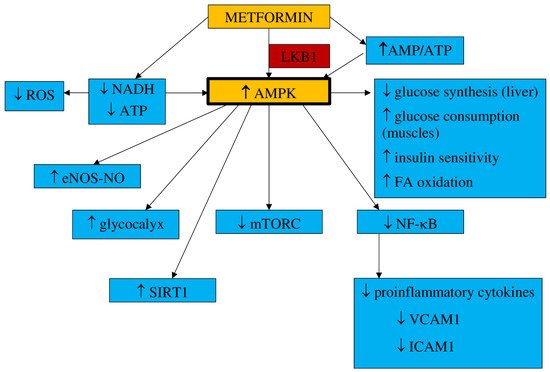
1.1. Pharmacokinetics and Mechanism of Action
Metformin is believed to be a safe medicine in pregnancy and is used primarily to treat women with gestational diabetes mellitus (GDM) [
81]. Metformin, a dimethyl-biguanide hydrochloride, is an oral hypoglycemic agent of a molecular weight of 129 daltons, absorbed within the duodenum and jejunum and excreted unchanged with urine and bile [
82]. The hypoglycemic effect of metformin is the result of several mechanisms, such as reduced gluconeogenesis in the liver, with limited hepatic glucose synthesis; decreased glucose absorption in the gastrointestinal tract; and intensification of its uptake in skeletal muscles [
83]. Its maximum dose is estimated to be 2.5–3 g daily (35–42 mg/kg) [
84]. It has been found that metformin acts within mitochondria, where it inhibits complex I of the mitochondrial electron transport chain (ETC), which leads to a reduction in nicotinamide adenine dinucleotide (NADH) oxidation and adenosine triphosphate (ATP) synthesis. It results in the activation of the 5′-adenosine monophosphate (AMP) kinase (AMPK), an increase in AMP concentrations, and the inhibition of the cAMP/PKA pathway (protein kinase A). Metformin activates the AMPK pathway, which results in a decrease in hepatic glucose synthesis and an increase in glucose consumption in muscles [
85,
86]. Additionally, metformin inhibits the transmembrane protein vATPase on the surface of the lysosome and raises the AMP/ATP ratio. It also stimulates AXIN-LKB1-vATPase and enhances AMPK protein attachment to the lysosome surface [
85]. The increased AMP/ATP ratio is responsible for the activation of AMPK, which results in suppressed glucose synthesis in the liver, enhanced insulin sensitivity and glucose uptake by muscle, and the activation of fatty acid oxidation [
87]. This process demands threonine 172 phosphorylation with liver kinase B1 (LKB1) [
88]. The AMPK pathway is triggered by metformin in a dose- and time-dependent manner. However, it has also been postulated that metformin can stimulate AMPK separately from AMP/ATP ratio variations [
89].
The results of animal studies have indicated that metformin may also decrease liver glucose synthesis concentrations without AMPK pathway involvement [
90]. According to the observations presented by Madiraju et al., gluconeogenesis can be restrained by metformin through mitochondrial glycerophosphate dehydrogenase suppression [
91]. AMPK activation influences cellular metabolism and cell growth and proliferation by blocking mTORC (mammalian target of rapamycin), which is a cancer-supporting pathway [
92]. Metformin may develop an antiproliferative effect by inhibiting mTORC1 through AMPK [
93,
94,
95].
The underlying mechanism by which metformin reduces the incidence of cardiovascular events and all-cause mortality has been actively investigated. It has been found that the induced AMPK pathway may act not only as a hypoglycemic and antiproliferative agent but also favorably influences the cardiovascular system. It may reduce inflammatory cell proliferation and their adhesion to the endothelium and lipid accumulation, and it is involved in the activation of genes responsible for cellular antioxidant defense and enzymes committed in the synthesis of nitric oxide [
96]. As a result, this translates into favorable effects on clinical signs and symptoms, such as hypertension, obesity and overweight, atherogenic dyslipidemia, procoagulant and thrombosis conditions, and carotid intima-media thickness, all of which are improved [
97,
98]. Metformin has been found to protect against cardiovascular complications, mainly by improving the function of the endothelium and by its anti-inflammatory properties, which have a positive impact on blood pressure, coagulation processes, and overweight/obesity [
99,
100].
Studies on animal models have proven that metformin can restore normal endothelium function [
101].
- a.
-
Inflammation and oxidative stress
There are many available reports on in vitro and animal models confirming the antiinflammatory properties of metformin. It has been suggested that metformin, by AMPK pathway activation, can restrain nuclear factor kappa B (NF-κB), which results in the limitation of proinflammatory gene expression [
102,
103]. NF-κB inhibition by metformin is also the effect of the blockade of the phosphoinositide 3-kinase (PI3K)-Akt pathway in human vascular wall cells [
104].
It has been observed that NF-κB suppression in macrophages, monocytes, and lymphocytes may finally result in a decrease in proinflammatory cytokines levels such as IL-1β, IL-6, and tumor necrosis factor-α (TNF-α), monocyte chemoattractant protein-1 (MCP-1), and IL-8, IL-2, and interferon as well as NO and prostaglandin E2 (PGE2) release [
105]. Gongol et al. found that metformin may inhibit the TNF-α–induced gene expression regulating E-selectin, vascular cell adhesion molecule 1 (VCAM1), intracellular adhesion molecule 1 (ICAM1), and MCP1 release. All of them contribute to monocyte adhesion to activated endothelial cells, suggesting that metformin could be a useful agent in preventing monocyte adhesion to endothelial cells [
106]. Thus, the influence on the NF-κB pathway may represent an interesting target for anti-inflammatory therapies. In addition, metformin has been shown to reduce the proinflammatory response by affecting AMPK-phosphatase and the tensin homolog (PTEN) [
107].
Metformin also diminishes the synthesis of advanced glycation end-products (AGEs), the levels of which increase due to hyperglycemia. AGEs have been found to exert proinflammatory properties and are believed to be one of the reasons for the development of vascular complications in diabetes mellitus [
108,
109]. AGEs have been revealed to induce oxidative stress as well as activate proinflammatory processes in the endothelium [
110]. Mamptu et al. observed that metformin inhibits the monocyte adhesion to the endothelium caused by AGEs [
111]. However, the exact mechanism of metformin action to reduce inflammation processes—directly or indirectly through glycemic normalization—has not been definitively established [
112].
It has been found that metformin, through the inhibition of nicotinamide adenine dinucleotide phosphate (NADPH), diminishes ROS production in endothelial cells [
113]. The results of the studies conducted by Bakhashab et al. indicate that metformin intensifies the expression of VEGFs responsible for the enhancement of angiogenesis in hypoxia and hyperglycemia conditions [
114].
The bioavailability of NO, a potent vasodilator, is one of the key factors in maintaining physiological endothelium properties and function. Metformin has been found to enhance the eNOS-NO pathway through the activation of AMPK in a dose-dependent manner. By this mechanism observed in endothelial cells in vitro, metformin may increase NO-mediated vasodilatation [
115].
- c.
-
Endothelial senescence and apoptosis
It has been claimed that hyperglycemia could be responsible for the senescence and apoptosis of endothelial cells that eventually lead to the loss of their function. Metformin, by significantly enhancing the expression of SIRT1, has been observed to reduce these processes and enable the endothelium to maintain its properties [
116]. SIRT1 has been shown to increase eNOS deacetylation and augment the bioavailability of NO, leading to a reduction of apoptosis and angiogenesis in the endothelium [
117].
Hyperglycemia is one of the factors increasing vascular permeability that finally result in endothelial leakage and the extravasation of monocytes, which is associate with impaired endothelial function. The endothelial glycocalyx, one of the matrix structures, prevents the increase in endothelial permeability. The results from animal model studies have shown that endothelial permeability is inhibited by metformin via AMPK activation, and the glycocalyx barrier is reinforced [
118].
The main mechanisms of metformin action are shown in Figure 1.
Figure 1. Main mechanisms of metformin action. AMPK—5′-adenosine monophosphate-activated protein kinase, AMP—5′-adenosine monophosphate, ATP—adenosine triphosphate, LKB1—liver kinase B1, NADH—nicotinamide adenine dinucleotide, ROS—reactive oxygen species, eNOS—endothelial nitric oxide synthase, NO—nitric oxide, SIRT1—sirtuin 1, mTORC—mammalian target of rapamycin, FA—fatty acids, NF-κB—nuclear factor kappa B, VCAM1—vascular cell adhesion molecule 1, ICAM1—intracellular adhesion molecule 1, MCP1—monocyte chemoattractant protein 1.
1.2. Impact on Preeclampsia Pathophysiology
Elevated insulin levels are believed to be exceptionally toxic to trophoblast cells in the first trimester of pregnancy and may be responsible for damage to their DNA, apoptosis, and limiting their survival. Hence, metformin use may prevent these events. These findings also suggest the need to consider screening for insulin resistance before conception to prevent hyperinsulinemia early in pregnancy [
119].
It has been suggested that the development of preeclampsia could be related to proinflammatory conditions that lead to the release of free radicals within the placenta and the consecutive oxidative/nitrosative stress [
120]. The results of the study conducted by Han et al. revealed that high glucose concentrations had a significant impact on the rise in trophoblast synthesis of proinflammatory cytokines such as IL-1β, IL-6, and IL-8 as well as the synthesis of antiangiogenic factors sFlt-1 and sEnd. They also reduce trophoblast migration. This may indicate the existence of a mechanism linking hyperglycemia to the development of PE and the role of metformin as a potential preventive agent. However, they also observed that metformin limited the glucose-induced inflammatory response moderately without any impact on the antiangiogenic or antimigratory response [
121]. This observation has been confirmed by Chiswick et al., who found that women treated with metformin during pregnancy had lower proinflammatory interleukin-6 levels [
122]. The metformin impact on inflammation and oxidative stress was examined in numerous animal and in vitro studies. Hu et al. observed that in a rat model of PE induced by lipopolysaccharides (LPS), metformin decreased the LPS-dependent secretion of proinflammatory cytokines such as TNF-α and IL-6 and limited oxidative/nitrative stress by enhancing the activity of superoxide dismutase (SOD). The placental NF-κB signaling pathway, activated by LPS, was suppressed. This resulted in a normalization of blood pressure, reduced proteinuria, improvement in fetal growth, and decreased stillbirth frequency. The authors concluded that metformin is beneficial to the PE-like rat model by protecting placentas from injury; thus, it could be an attractive agent for PE prevention and/or treatment [
123]. It has been reported that the decrease in IL-27, TNF-α, and IL-6 expression in vivo (in both preeclamptic rat models and trophoblast cells) was the result of H19 inhibition by metformin in a dose-dependent manner [
124]. On the other hand, Correia-Branco et al. showed the adverse influence of metformin on an extravillous trophoblastic cell line, with reducing cell proliferation rates, culture growth, viability, and capacity of migration. Thus, they were of the opinion that the processes involved in placentation could be highly impaired by metformin, with mTORC and PI3K involvement [
125].
Metformin acts as an endothelial protective agent via the AMPK activation pathway not only in diabetic patients but also in healthy individuals in a glucose-independent manner [
126]. The endothelium dysfunction reported in PE is correlated with an increase in the expression of ICAM1 and VICAM1, which is enhanced by proinflammatory cytokine TNFα [
127,
128]. Brownfoot et al. revealed that metformin diminished the VCAM1 levels induced by TNF-α in HUVECs (human umbilical vein endothelial cells) [
40,
129]. An abnormal invasion of the trophoblast leads to ischemia and hypoxia of the placenta and an increase in the concentrations of circulating vasoactive factors. Antiangiogenic factors such as soluble fms-like tyrosine kinase-1 and soluble endoglin cause imbalances in pro- and antiangiogenic factors [
130]. The possibility of restoring the balance by suppressing antiangiogenic agents seems attractive as a method of effective prophylaxis of preeclampsia. The results of the study of Brownfoot et al., conducted on endothelial cells, villous cytotrophoblast cells, and preterm preeclamptic placental villous explants, suggest that metformin in a dose-dependent manner decreases the synthesis of sFlt-1 and sENG. It also reversed the endothelial dysfunction observed in preeclampsia. The authors were of the opinion that the metformin effect was likely to be regulated at the mitochondrial level, probably by inhibiting the mitochondrial electron transport chain. In the same study, it has also been observed that the defective angiogenesis caused by sFlt-1 was improved with metformin. The authors of this research concluded that since metformin limited endothelium dysfunction, reinforced vasodilatation, and promoted angiogenesis, it might be useful for the prophylaxis or treatment of preeclampsia [
40]. This group of researchers also studied the effect of metformin and esmoprazol belonging to proton pump inhibitors and metformin and sulfasalazine combined on sFlt-1 mRNA expression and sFlt-1 secretion as well as sENG secretion. They have found metformin and esmoprazol to be more effective in decreasing sFlt-1 synthesis, with no additive impact on sENG levels compared to metformin alone [
129,
131]. The concomitant use of metformin with sulfasalazine resulted in diminished sFlt-1 and sENG release and enhanced VEGF alpha expression in cytotrophoblasts [
131].
According to the results of the abovementioned studies, their authors concluded that the combined use of metformin with esmoprazol or sulfasalazine seemed to be more effective in PE prophylaxis and treatment than metformin alone [
129,
131].
The increase in VEGF release induced by metformin has been revealed in numerous reports. VEGF represents the family of angiogenic factors participating in the development of placental vasculature and appropriate trophoblast invasion and implantation [
132]. It has been found that their levels are decreased in preeclampsia [
133]. An animal model study showed that metformin enhances VEGF synthesis and, consequently, improves angiogenesis in the placenta [
134].
The results of numerous studies have indicated that endothelial function may also be improved as a result of the action of other mechanisms that are modulated by metformin. NO is the leading vasodilator involved in cytotrophoblast invasion, implantation, and providing the development of low-resistance placental blood flow. Since impaired NO bioavailability and signaling have been reported in preeclampsia, a drug that can restore the balance in the NO pathway may be attractive for PE prophylaxis [
49]. Metformin has been found to raise NO synthesis through the activation of AMPK, which leads to the activation of eNOS [
115].
The results of studies conducted in diabetic patients have indicated that metformin stimulates a marked increase in the number of EPCs and strengthens angiogenic potential by activating the AMPK/eNOS pathway [
115,
135]. Asadian et al. presented the opposite opinion: they have shown no metformin impact on the number and activity of EPCs [
136]. There are also study results that have indicated the adverse effects of metformin on both the number and bioactivity of EPCs. It has been noticed that metformin suppresses the angiogenic capacity of EPCs and their migration [
137,
138]. The ambiguous conclusions of the research presented above may be the result of different doses of metformin and the small size of the study groups. Hence, it seems that drugs that have a beneficial effect on EPCs might be useful in the prevention or treatment of preeclampsia.
Endoplasmic reticulum stress is believed to be involved in the pathogenesis of preeclampsia by promoting the release of proinflammatory cytokines, antiangiogenic factors, and trophoblastic apoptotic debris [
57,
58,
59]. The results of the study of Suzuki et al., conducted in trophoblast-like cells, indicated that metformin, by limiting ERS, restored normal levels of PIGF, which might justify its use in the prevention of PE [
139].
Placentation, which is impaired in preeclampsia, requires extracellular matrix degradation with the involvement of metalloproteinases [
61]. Wang et al., conducting a study on the effect of metformin on PE-like animal models, have found that it improved vascularization and contributed to an increase in the concentration of MMP-2 and VEGF in preeclamptic placental tissue [
134].
Additionally, metformin, by inducing SIRT1 expression, is believed to significantly increase cell viability, decrease cell apoptosis, and reduce the release of proinflammatory cytokines, which allow the maintenance of physiological endothelium function [
72]. There are few reports on SIRT1 in preeclampsia, and data on the metformin effect on SIRT1 in preeclampsia are, so far, unavailable.
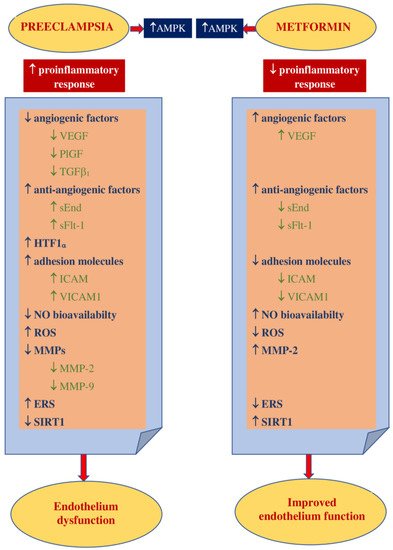
Figure 2 shows the main theoretical basis for the use of metformin in PE prophylaxis.
Figure 2. Theoretical basis for the use of metformin in PE prevention. AMPK—5’ adenosine monophosphate-activated protein kinase, VEGF—vascular endothelial growth factor, PlGF—placental growth factor, TGF β1—transforming growth factor-β1, sEnd—soluble endoglin, sFlt-1—fms-like tyrosine kinase-1, HTF1 α—hypoxia-inducible factor 1α, ICAM1—intracellular cell adhesion molecule 1, VICAM1—vascular cell adhesion molecule 1, NO—nitric oxide, ROS—reactive oxygen species, MMPs—matrix metalloproteinases, MMP-2—matrix metalloproteinase-2, MMP-9—matrix metalloproteinase-9, ERS—endoplasmic reticulum stress, EPCs—endothelial progenitor cells, SIRT1—sirtuin1.