The possibility of prophylaxis of hypertensive disorders of pregnancy (HDPs) such as preeclampsia (PE) and pregnancy-induced hypertension (PIH) is of interest due to the unpredictable course of these diseases and the risks they carry for both mother and fetus. It has been proven that their development is associated with the presence of the placenta, and the processes that initiate it begin at the time of the abnormal invasion of the trophoblast in early pregnancy. The ideal HDPs prophylaxis should alleviate the influence of risk factors and, at the same time, promote physiological trophoblast invasion and maintain the physiologic endothelium function without any harm to both mother and fetus. So far, aspirin is the only effective and recommended pharmacological agent for the prevention of HDPs in high-risk groups. Metformin is a hypoglycemic drug with a proven protective effect on the cardiovascular system. Respecting the anti-inflammatory properties of metformin and its favorable impact on the endothelium, it seems to be an interesting option for HDPs prophylaxis.
- preeclampsia
- pregnancy-induced hypertension
- metformin
- pregnancy
1. Metformin
1.1. Pharmacokinetics and Mechanism of Action
- a.
-
Inflammation and oxidative stressThere are many available reports on in vitro and animal models confirming the antiinflammatory properties of metformin. It has been suggested that metformin, by AMPK pathway activation, can restrain nuclear factor kappa B (NF-κB), which results in the limitation of proinflammatory gene expression [102,103][22][23]. NF-κB inhibition by metformin is also the effect of the blockade of the phosphoinositide 3-kinase (PI3K)-Akt pathway in human vascular wall cells [104][24].It has been observed that NF-κB suppression in macrophages, monocytes, and lymphocytes may finally result in a decrease in proinflammatory cytokines levels such as IL-1β, IL-6, and tumor necrosis factor-α (TNF-α), monocyte chemoattractant protein-1 (MCP-1), and IL-8, IL-2, and interferon as well as NO and prostaglandin E2 (PGE2) release [105][25]. Gongol et al. found that metformin may inhibit the TNF-α–induced gene expression regulating E-selectin, vascular cell adhesion molecule 1 (VCAM1), intracellular adhesion molecule 1 (ICAM1), and MCP1 release. All of them contribute to monocyte adhesion to activated endothelial cells, suggesting that metformin could be a useful agent in preventing monocyte adhesion to endothelial cells [106][26]. Thus, the influence on the NF-κB pathway may represent an interesting target for anti-inflammatory therapies. In addition, metformin has been shown to reduce the proinflammatory response by affecting AMPK-phosphatase and the tensin homolog (PTEN) [107][27].Metformin also diminishes the synthesis of advanced glycation end-products (AGEs), the levels of which increase due to hyperglycemia. AGEs have been found to exert proinflammatory properties and are believed to be one of the reasons for the development of vascular complications in diabetes mellitus [108,109][28][29]. AGEs have been revealed to induce oxidative stress as well as activate proinflammatory processes in the endothelium [110][30]. Mamptu et al. observed that metformin inhibits the monocyte adhesion to the endothelium caused by AGEs [111][31]. However, the exact mechanism of metformin action to reduce inflammation processes—directly or indirectly through glycemic normalization—has not been definitively established [112][32].It has been found that metformin, through the inhibition of nicotinamide adenine dinucleotide phosphate (NADPH), diminishes ROS production in endothelial cells [113][33]. The results of the studies conducted by Bakhashab et al. indicate that metformin intensifies the expression of VEGFs responsible for the enhancement of angiogenesis in hypoxia and hyperglycemia conditions [114][34].
- b.
-
NO synthesisThe bioavailability of NO, a potent vasodilator, is one of the key factors in maintaining physiological endothelium properties and function. Metformin has been found to enhance the eNOS-NO pathway through the activation of AMPK in a dose-dependent manner. By this mechanism observed in endothelial cells in vitro, metformin may increase NO-mediated vasodilatation [115][35].
- c.
-
Endothelial senescence and apoptosisIt has been claimed that hyperglycemia could be responsible for the senescence and apoptosis of endothelial cells that eventually lead to the loss of their function. Metformin, by significantly enhancing the expression of SIRT1, has been observed to reduce these processes and enable the endothelium to maintain its properties [116][36]. SIRT1 has been shown to increase eNOS deacetylation and augment the bioavailability of NO, leading to a reduction of apoptosis and angiogenesis in the endothelium [117][37].
- d.
-
Vascular integrityHyperglycemia is one of the factors increasing vascular permeability that finally result in endothelial leakage and the extravasation of monocytes, which is associate with impaired endothelial function. The endothelial glycocalyx, one of the matrix structures, prevents the increase in endothelial permeability. The results from animal model studies have shown that endothelial permeability is inhibited by metformin via AMPK activation, and the glycocalyx barrier is reinforced [118][38].The main mechanisms of metformin action are shown in Figure 1.
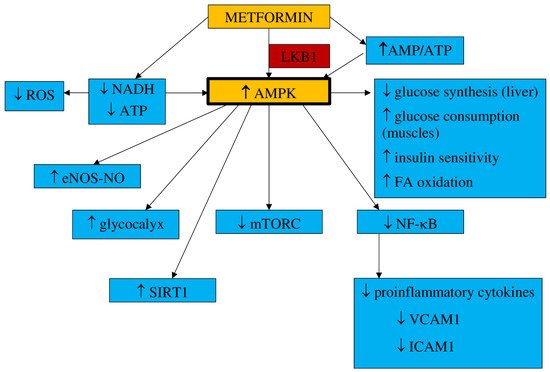 Figure 1. Main mechanisms of metformin action. AMPK—5′-adenosine monophosphate-activated protein kinase, AMP—5′-adenosine monophosphate, ATP—adenosine triphosphate, LKB1—liver kinase B1, NADH—nicotinamide adenine dinucleotide, ROS—reactive oxygen species, eNOS—endothelial nitric oxide synthase, NO—nitric oxide, SIRT1—sirtuin 1, mTORC—mammalian target of rapamycin, FA—fatty acids, NF-κB—nuclear factor kappa B, VCAM1—vascular cell adhesion molecule 1, ICAM1—intracellular adhesion molecule 1, MCP1—monocyte chemoattractant protein 1.
Figure 1. Main mechanisms of metformin action. AMPK—5′-adenosine monophosphate-activated protein kinase, AMP—5′-adenosine monophosphate, ATP—adenosine triphosphate, LKB1—liver kinase B1, NADH—nicotinamide adenine dinucleotide, ROS—reactive oxygen species, eNOS—endothelial nitric oxide synthase, NO—nitric oxide, SIRT1—sirtuin 1, mTORC—mammalian target of rapamycin, FA—fatty acids, NF-κB—nuclear factor kappa B, VCAM1—vascular cell adhesion molecule 1, ICAM1—intracellular adhesion molecule 1, MCP1—monocyte chemoattractant protein 1.1.2. Impact on Preeclampsia Pathophysiology
Elevated insulin levels are believed to be exceptionally toxic to trophoblast cells in the first trimester of pregnancy and may be responsible for damage to their DNA, apoptosis, and limiting their survival. Hence, metformin use may prevent these events. These findings also suggest the need to consider screening for insulin resistance before conception to prevent hyperinsulinemia early in pregnancy [119][39].It has been suggested that the development of preeclampsia could be related to proinflammatory conditions that lead to the release of free radicals within the placenta and the consecutive oxidative/nitrosative stress [120][40]. The results of the study conducted by Han et al. revealed that high glucose concentrations had a significant impact on the rise in trophoblast synthesis of proinflammatory cytokines such as IL-1β, IL-6, and IL-8 as well as the synthesis of antiangiogenic factors sFlt-1 and sEnd. They also reduce trophoblast migration. This may indicate the existence of a mechanism linking hyperglycemia to the development of PE and the role of metformin as a potential preventive agent. However, they also observed that metformin limited the glucose-induced inflammatory response moderately without any impact on the antiangiogenic or antimigratory response [121][41]. This observation has been confirmed by Chiswick et al., who found that women treated with metformin during pregnancy had lower proinflammatory interleukin-6 levels [122][42]. The metformin impact on inflammation and oxidative stress was examined in numerous animal and in vitro studies. Hu et al. observed that in a rat model of PE induced by lipopolysaccharides (LPS), metformin decreased the LPS-dependent secretion of proinflammatory cytokines such as TNF-α and IL-6 and limited oxidative/nitrative stress by enhancing the activity of superoxide dismutase (SOD). The placental NF-κB signaling pathway, activated by LPS, was suppressed. This resulted in a normalization of blood pressure, reduced proteinuria, improvement in fetal growth, and decreased stillbirth frequency. The authors concluded that metformin is beneficial to the PE-like rat model by protecting placentas from injury; thus, it could be an attractive agent for PE prevention and/or treatment [123][43]. It has been reported that the decrease in IL-27, TNF-α, and IL-6 expression in vivo (in both preeclamptic rat models and trophoblast cells) was the result of H19 inhibition by metformin in a dose-dependent manner [124][44]. On the other hand, Correia-Branco et al. showed the adverse influence of metformin on an extravillous trophoblastic cell line, with reducing cell proliferation rates, culture growth, viability, and capacity of migration. Thus, they were of the opinion that the processes involved in placentation could be highly impaired by metformin, with mTORC and PI3K involvement [125][45].Metformin acts as an endothelial protective agent via the AMPK activation pathway not only in diabetic patients but also in healthy individuals in a glucose-independent manner [126][46]. The endothelium dysfunction reported in PE is correlated with an increase in the expression of ICAM1 and VICAM1, which is enhanced by proinflammatory cytokine TNFα [127,128][47][48]. Brownfoot et al. revealed that metformin diminished the VCAM1 levels induced by TNF-α in HUVECs (human umbilical vein endothelial cells) [40,129][49][50]. An abnormal invasion of the trophoblast leads to ischemia and hypoxia of the placenta and an increase in the concentrations of circulating vasoactive factors. Antiangiogenic factors such as soluble fms-like tyrosine kinase-1 and soluble endoglin cause imbalances in pro- and antiangiogenic factors [130][51]. The possibility of restoring the balance by suppressing antiangiogenic agents seems attractive as a method of effective prophylaxis of preeclampsia. The results of the study of Brownfoot et al., conducted on endothelial cells, villous cytotrophoblast cells, and preterm preeclamptic placental villous explants, suggest that metformin in a dose-dependent manner decreases the synthesis of sFlt-1 and sENG. It also reversed the endothelial dysfunction observed in preeclampsia. The authors were of the opinion that the metformin effect was likely to be regulated at the mitochondrial level, probably by inhibiting the mitochondrial electron transport chain. In the same study, it has also been observed that the defective angiogenesis caused by sFlt-1 was improved with metformin. The authors of this research concluded that since metformin limited endothelium dysfunction, reinforced vasodilatation, and promoted angiogenesis, it might be useful for the prophylaxis or treatment of preeclampsia [40][49]. This group of researchers also studied the effect of metformin and esmoprazol belonging to proton pump inhibitors and metformin and sulfasalazine combined on sFlt-1 mRNA expression and sFlt-1 secretion as well as sENG secretion. They have found metformin and esmoprazol to be more effective in decreasing sFlt-1 synthesis, with no additive impact on sENG levels compared to metformin alone [129,131][50][52]. The concomitant use of metformin with sulfasalazine resulted in diminished sFlt-1 and sENG release and enhanced VEGF alpha expression in cytotrophoblasts [131][52].According to the results of the abovementioned studies, their authors concluded that the combined use of metformin with esmoprazol or sulfasalazine seemed to be more effective in PE prophylaxis and treatment than metformin alone [129,131][50][52].The increase in VEGF release induced by metformin has been revealed in numerous reports. VEGF represents the family of angiogenic factors participating in the development of placental vasculature and appropriate trophoblast invasion and implantation [132][53]. It has been found that their levels are decreased in preeclampsia [133][54]. An animal model study showed that metformin enhances VEGF synthesis and, consequently, improves angiogenesis in the placenta [134][55].The results of numerous studies have indicated that endothelial function may also be improved as a result of the action of other mechanisms that are modulated by metformin. NO is the leading vasodilator involved in cytotrophoblast invasion, implantation, and providing the development of low-resistance placental blood flow. Since impaired NO bioavailability and signaling have been reported in preeclampsia, a drug that can restore the balance in the NO pathway may be attractive for PE prophylaxis [49][56]. Metformin has been found to raise NO synthesis through the activation of AMPK, which leads to the activation of eNOS [115][35].The results of studies conducted in diabetic patients have indicated that metformin stimulates a marked increase in the number of EPCs and strengthens angiogenic potential by activating the AMPK/eNOS pathway [115,135][35][57]. Asadian et al. presented the opposite opinion: they have shown no metformin impact on the number and activity of EPCs [136][58]. There are also study results that have indicated the adverse effects of metformin on both the number and bioactivity of EPCs. It has been noticed that metformin suppresses the angiogenic capacity of EPCs and their migration [137,138][59][60]. The ambiguous conclusions of the research presented above may be the result of different doses of metformin and the small size of the study groups. Hence, it seems that drugs that have a beneficial effect on EPCs might be useful in the prevention or treatment of preeclampsia.Endoplasmic reticulum stress is believed to be involved in the pathogenesis of preeclampsia by promoting the release of proinflammatory cytokines, antiangiogenic factors, and trophoblastic apoptotic debris [57,58,59][61][62][63]. The results of the study of Suzuki et al., conducted in trophoblast-like cells, indicated that metformin, by limiting ERS, restored normal levels of PIGF, which might justify its use in the prevention of PE [139][64].Placentation, which is impaired in preeclampsia, requires extracellular matrix degradation with the involvement of metalloproteinases [61][65]. Wang et al., conducting a study on the effect of metformin on PE-like animal models, have found that it improved vascularization and contributed to an increase in the concentration of MMP-2 and VEGF in preeclamptic placental tissue [134][55].Additionally, metformin, by inducing SIRT1 expression, is believed to significantly increase cell viability, decrease cell apoptosis, and reduce the release of proinflammatory cytokines, which allow the maintenance of physiological endothelium function [72][66]. There are few reports on SIRT1 in preeclampsia, and data on the metformin effect on SIRT1 in preeclampsia are, so far, unavailable.Figure 2 shows the main theoretical basis for the use of metformin in PE prophylaxis.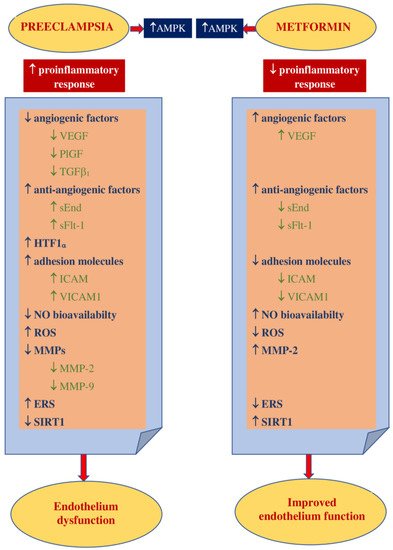 Figure 2. Theoretical basis for the use of metformin in PE prevention. AMPK—5’ adenosine monophosphate-activated protein kinase, VEGF—vascular endothelial growth factor, PlGF—placental growth factor, TGF β1—transforming growth factor-β1, sEnd—soluble endoglin, sFlt-1—fms-like tyrosine kinase-1, HTF1 α—hypoxia-inducible factor 1α, ICAM1—intracellular cell adhesion molecule 1, VICAM1—vascular cell adhesion molecule 1, NO—nitric oxide, ROS—reactive oxygen species, MMPs—matrix metalloproteinases, MMP-2—matrix metalloproteinase-2, MMP-9—matrix metalloproteinase-9, ERS—endoplasmic reticulum stress, EPCs—endothelial progenitor cells, SIRT1—sirtuin1.
Figure 2. Theoretical basis for the use of metformin in PE prevention. AMPK—5’ adenosine monophosphate-activated protein kinase, VEGF—vascular endothelial growth factor, PlGF—placental growth factor, TGF β1—transforming growth factor-β1, sEnd—soluble endoglin, sFlt-1—fms-like tyrosine kinase-1, HTF1 α—hypoxia-inducible factor 1α, ICAM1—intracellular cell adhesion molecule 1, VICAM1—vascular cell adhesion molecule 1, NO—nitric oxide, ROS—reactive oxygen species, MMPs—matrix metalloproteinases, MMP-2—matrix metalloproteinase-2, MMP-9—matrix metalloproteinase-9, ERS—endoplasmic reticulum stress, EPCs—endothelial progenitor cells, SIRT1—sirtuin1.
2. Metformin in Preventing Hypertensive Disorders of Pregnancy
Due to the effect of metformin, far beyond the impact on carbohydrate metabolism, it becomes an attractive drug for the prevention of hypertensive disorders of pregnancy. The studies published so far have focused mainly on its effects on pregnancy outcomes and the fetus and child and were conducted primarily in women with gestational diabetes (GDM), polycystic ovary syndrome (PCOS), and obesity. The main objective of these studies was not to assess the effect of metformin on the development of hypertensive complications in pregnancy. This chapter presents the results of randomized controlled trials (RCTs) and meta-analyses that have been published over the past 10 years. Electronic databases Pubmed and MEDLINE were searched using keywords such as metformin and pregnancy. Only articles available in English were taken into account. Of the 110 published RCTs, only 10 provided information on the effect of metformin on preeclampsia and/or pregnancy-induced hypertension incidences, and 11 out of 74 meta-analyses did. The results of the selected randomized controlled trials that have been published within the last 10 years and provide information on metformin influence on the frequency of preeclampsia are presented in Table 1. Some analyses have also taken into account the incidence of pregnancy-induced hypertension or gestational hypertension; these terms were used interchangeably for gestational hypertension. In none of the following work metformin impact on the incidence of hypertensive complications of pregnancy was the primary outcome.| Studied Group | Size of Groups | Metformin Dose | GA at Entry to the Study | PIH and PE | Authors | ||
|---|---|---|---|---|---|---|---|
| GDM high risk | SG: metformin—24 CG:no treatment—25 |
500–1000 mg | 14th week | PE SG: 0% (0) CG: 8.7% (2) p = 0.049 |
Brink et al., 2018 [67] | ||
| GDM | SG: metformin—43 CG:insulin alone—57 |
500–2500 mg | 20th–36th week | Obesity | metformin vs. placebo | 840PIH SG: 18.6% (8) CG: 24% (18) NS PE SG: 0% (0) CG: 8% (6) p = 0.05 |
Ainuddin et al., 2014 [68] |
| GDM | SG: metformin 110 CG insulin 107 |
500–2000 mg | 22nd–34th week | ||||
| GDM | Metformin vs. insulin | 2165 | ↓PIH RR 0.56 95% CI 0.37–0.85PIH SG: 1.8% (2) CG: 3.7% (4) p = 0.42, RR 0.5 95% CI 0.1–2.7 PE SG: 4.6% (5) CG: 9.4% (10) p = 0.19, RR 0.5 95% CI 0.2–1.4 |
Butalia et al., 2017 [81]Tertti et al., 2013 [69] | |||
| GDM | SG metformin: 86 CG insulin: 80 |
1000–2500 mg | 20th–34th week | PIH | |||
| GDM | metformin vs. insulin | 1556 | ↓HDPs RR 0.82 95% CI 0.67–1.0 SG: 5% (4) CG 13.8% (11) p = 0.058, RR 0.4 95% CI 0.1–1.1 PE SG: 6.3% (5) CG: 8.8% (7) p = 0.548, RR 0.7 95% CI 0.2–2.22 |
Feng et al., 2017 [82Niromanesh et al., 2012 [70] | |||
| ] | DMt.2 | SG: metformin—233 CG: insulin—240 |
2000 mg | 6th–22th week | |||
| GDM | metformin vs. insulin | 1110 | ↓PIH RR 0.53 95% CI 0.31–0.90 PE RR 0.81 95% CI 0.55–1.17, | PIH SG: 5% (13) CG 6% (15) p = 0.82, RR 0.92 95% CI 0.46–1.8. PE SG: 15% (37) CG 12% (30) p = 0.29, RR 1.27 95% CI 0.82–1.92 Chronic HT ST: 8% (20) CG: 9% (22) p = 0.68, RR 0.89 95% CI 0.51–1.56 |
Feig et al., 2020 [71] | ||
| DMt.2 | SG: metformin alone—16 CG: insulin alone 100 |
500–2500 mg | about 10th week | PIH SG: 6.2% (1) CG: 36% (36) p= 0.020 PE SG: 25% (4) CG: 17% (17) p = 0.084 |
Ainuddin et al., 2015 [72] | ||
| Obesity | SG: metformin—171 CG: placebo—186 |
1000 mg | <20th week | PE SG: 3.5% (6) CG: 4.8% (9) p = 0.01, RR 0.17 95% CI 0.10–1.41 |
Nascimento et al., 2020 [73] | ||
| Obesity (35 kg/m2) | SG: metformin—202 CG: placebo—198 |
1000–3000 mg | 12th–18th week | PIH SG: 6.4% (13) CG: 6.7% (13) p = 0.93, RR 0.96 95% CI 0.43–2.13 PE SG: 3% (6) CG: 11.3% (13) p = 0.001, RR 0.24 95% CI 0.10–0.61 |
Syngelaki et al., 2016 [74] | ||
| Obesity (BMI > 30 kg/m2) | SG: metformin—221 CG: placebo—222 |
500–2500 mg | 12th–16th week | PIH SG: 10% (21) CG 6% (14) p= 0.22, RR 1.56 95% CI 0.77–3.15. PE SG: 3% (7) CG 1% (3) p = 0.21, RR 2.39 95% 0.61–9.36 |
Chiswick et al., 2015 [42] | ||
| PCOS | SG: metformin—238 CG: placebo—240 |
1000–2000 mg | in the 1st trimester | PE SG: 3% (8) CG: 7% (17) p = 0.10, RR 0.46 95% CI 0.17–1.15 |
Løvvik et al., 2019 [75] |
| Studied Group | Comparison | Number of Participants | Metformin Impact on PIH/PE | Authors |
|---|---|---|---|---|
| GDM | metformin vs. insulin | 1260 | PIH RR 0.56 95% CI 0.37–0.85 PE RR 0.83 95% CI 0.60–1.14 PE RR 0.74 95% CI 0.09–6.28 |
Kalafat et al., 2018 [80] |
| Li et al., 2015 | [ | 83 | ] | |
| 1634 | ||||
| GDM | metformin vs. insulin | 1110 | ↓PIH RR 0.55 95% CI 0.31–0.91 PE RR 0.84 95% CI 0.57–1.23 |
Poolsup et al., 2014 [84] |
| 1299 | ||||
| GDM | metformin vs. insulin | 1712 | PE RR = 0.82 95% CI 0.56–1.2 |
Zhu et al., 2014 [85] |
| GDM | metformin vs. insulin | 1110 | ↓PIH RR 0.52 95%CI 0.30–0.90 |
Gui et al., 2013 [86] |
| Obesity | metformin vs. no-treatment | 840 614 308 |
PIH (obesity) RR 1.24 95% CI 0.76–2.02 ↓PE (obesity) RR 0.51 95% CI 0.26–0.98 ↓PIH (PCOS) RR 0.37 95% CI 0.25–0.57 PE (PCOS) RR 1.96 95% CI 0.81–4.77 ↓PIH (GDM) RR 0.53 95% CI 0.31–0.90 PE (GDM) RR 0.70 95% CI 0.45–1.10 |
Nascimento et al., 2018 [79] |
| PCOS | ||||
| GDM | metformin vs. insulin | 1120 1120 |
||
| Obesity | metformin vs. no-treatment or placebo | 1034 | PIH RR 1.02 95% CI 0.54–1.94 PE RR 0.74 95% CI 0.09–6.28 |
Dodd et al., 2018 [87] |
| PCOS | metformin vs. no-treatment or placebo | 929 | PE RR 0.92 95% CI 0.28–3.00 |
Feng et al., 2015 [88] |
| PCOS | metformin vs. no-treatment or placebo | 878 | ↓PE RR 0.53 95% CI 0.30–0.95 |
Zheng et al., 2013 [89] |
References
- Rowan, J.A.; Hague, W.M.; Gao, W.; Battin, M.R.; Moore, M.P. MiG Trial Investigators. Metformin versus insulin for the treatment of gestational diabetes. N. Engl. J. Med. 2008, 358, 2003–2015.
- Graham, G.G.; Punt, J.; Arora, M.; Day, R.O.; Doogue, M.P.; Duong, J.K.; Furlong, T.J.; Greenfield, J.R.; Greenup, L.C.; Kirkpatrick, C.M.; et al. Clinical pharmacokinetics of metformin. Clin. Pharmacokinet. 2011, 50, 81–98.
- Foretz, M.; Guigas, B.; Bertrand, L.; Pollak, M.; Viollet, B. Metformin: From mechanisms of action to therapies. Cell Metab. 2014, 20, 953–966.
- He, L.; Wondisford, F.E. Metformin action: Concentrations matter. Cell Metab. 2015, 21, 159–162.
- El-Mir, M.Y.; Nogueira, V.; Fontaine, E.; Avéret, N.; Rigoulet, M.; Leverve, X. Dimethylbiguanide inhibits cell respiration via an indirect effect targeted on the respiratory chain complex I. J. Biol. Chem. 2000, 275, 223–228.
- Oliveras-Ferraros, C.; Vazquez-Martin, A.; Menendez, J.A. Genome-wide inhibitory impact of the AMPK activator metformin on [kinesins, tubulins, histones, auroras and polo-like kinases] M-phase cell cycle genes in human breast cancer cells. Cell Cycle 2009, 8, 1633–1636.
- Zhou, G.; Myers, R.; Li, Y.; Chen, Y.; Shen, X.; Fenyk-Melody, J.; Wu, M.; Ventre, J.; Doebber, T.; Fujii, N.; et al. Role of AMP-activated protein kinase in mechanism of metformin action. J. Clin. Investig. 2001, 108, 1167–1174.
- Shaw, R.J.; Lamia, K.A.; Vasquez, D.; Koo, S.H.; Bardeesy, N.; Depinho, R.A.; Montminy, M.; Cantley, L.C. The kinase LKB1 mediates glucose homeostasis in liver and therapeutic effects of metformin. Science 2005, 310, 1642–1646.
- Zou, M.H.; Kirkpatrick, S.S.; Davis, B.J.; Nelson, J.S.; Wiles, W.G., IV; Schlattner, U.; Neumann, D.; Brownlee, M.; Freeman, M.B.; Goldman, M.H. Activation of the AMP-activated protein kinase by the anti-diabetic drug metformin in vivo. Role of mitochondrial reactive nitrogen species. J. Biol. Chem. 2004, 279, 43940–43951.
- Foretz, M.; Hébrard, S.; Leclerc, J.; Zarrinpashneh, E.; Soty, M.; Mithieux, G.; Sakamoto, K.; Andreelli, F.; Viollet, B. Metformin inhibits hepatic gluconeogenesis in mice independently of the LKB1/AMPK pathway via a decrease in hepatic energy state. J. Clin. Investig. 2010, 120, 2355–2369.
- Madiraju, A.K.; Erion, D.M.; Rahimi, Y.; Zhang, X.M.; Braddock, D.T.; Albright, R.A.; Prigaro, B.J.; Wood, J.L.; Bhanot, S.; MacDonald, M.J.; et al. Metformin suppresses gluconeogenesis by inhibiting mitochondrial glycerophosphate dehydrogenase. Nature 2014, 510, 542–546.
- Luo, Z.; Zang, M.; Guo, W. AMPK as a metabolic tumor suppressor: Control of metabolism and cell growth. Future Oncol. 2010, 6, 457–470.
- Bost, F.; Sahra, I.B.; Le Marchand-Brustel, Y.; Tanti, J.F. Metformin and cancer therapy. Curr. Opin. Oncol. 2012, 24, 103–108.
- Dowling, R.J.; Zakikhani, M.; Fantus, I.G.; Pollak, M.; Sonenberg, N. Metformin inhibits mammalian target of rapamycin-dependent translation initiation in breast cancer cells. Cancer Res. 2007, 67, 10804108012.
- Ben Sahra, I.; Regazzetti, C.; Robert, G.; Laurent, K.; Le Marchand-Brustel, Y.; Auberger, P.; Tanti, J.F.; Giorgetti-Peraldi, S.; Bost, F. Metformin, independent of AMPK, induces mTOR inhibition and cell-cycle arrest through REDD1. Cancer Res. 2011, 71, 4366–4372.
- Scheen, A.J.; Esser, N.; Paquot, N. Antidiabetic agents: Potential anti-inflammatory activity beyond glucose control. Diabetes Metab. 2015, 41, 183–194.
- Saenz, A.; Fernandez-Esteban, I.; Mataix, A.; Ausejo, M.; Roque, M.; Moher, D. Metformin monotherapy for type 2 diabetes mellitus. Cochrane Database Syst. Rev. 2005, 3, CD002966.
- Abbasi, F.; Chu, J.W.; McLaughlin, T.; Lamendola, C.; Leary, E.T.; Reaven, G.M. Effect of metformin treatment on multiple cardiovascular disease risk factors in patients with type 2 diabetes mellitus. Metabolism 2004, 53, 159–164.
- De Aguiar, L.G.; Bahia, L.R.; Villela, N.; Laflor, C.; Sicuro, F.; Wiernsperger, N.; Bottino, D.; Bouskela, E. Metformin improves endothelial vascular reactivity in first-degree relatives of type 2 diabetic patients with metabolic syndrome and normal glucose tolerance. Diabetes Care 2006, 29, 1083–1089.
- Libby, P. Mechanisms of acute coronary syndromes and their implications for therapy. N. Engl. J. Med. 2013, 368, 2004–2013.
- Sena, C.M.; Matafome, P.; Louro, T.; Nunes, E.; Fernandes, R.; Seiça, R.M. Metformin restores endothelial function in aorta of diabetic rats. Br. J. Pharmacol. 2011, 163, 424–437.
- Li, S.N.; Wang, X.; Zeng, Q.T.; Feng, Y.B.; Cheng, X.; Mao, X.B.; Wang, T.H.; Deng, H.P. Metformin inhibits nuclear factor kappaB activation and decreases serum high-sensitivity C-reactive protein level in experimental atherogenesis of rabbits. Heart Vessels. 2009, 24, 446–453.
- Hattori, Y.; Suzuki, K.; Hattori, S.; Kasai, K. Metformin inhibits cytokine-induced nuclear factor kappaB activation via AMP-activated protein kinase activation in vascular endothelial cells. Hypertension 2006, 47, 1183–1888.
- Isoda, K.; Young, J.L.; Zirlik, A.; MacFarlane, L.A.; Tsuboi, N.; Gerdes, N.; Schönbeck, U.; Libby, P. Metformin inhibits proinflammatory responses and nuclear factor-kappaB in human vascular wall cells. Arterioscler. Thromb. Vasc. Biol. 2006, 26, 611–617.
- Hyun, B.; Shin, S.; Lee, A.; Lee, S.; Song, Y.; Ha, N.J.; Cho, K.H.; Kim, K. Metformin down-regulates TNF-α secretion via suppression of scavenger receptors in macrophages. Immune Netw. 2013, 13, 123–132.
- Gongol, B.; Marin, T.; Peng, I.C.; Woo, B.; Martin, M.; King, S.; Sun, W.; Johnson, D.A.; Chien, S.; Shyy, J.Y. AMPKα2 exerts its anti-inflammatory effects through PARP-1 and Bcl-6. Proc. Natl. Acad. Sci. USA 2013, 110, 3161–3166.
- Kim, S.A.; Choi, H.C. Metformin inhibits inflammatory response via AMPK-PTEN pathway in vascular smooth muscle cells. Biochem. Biophys. Res. Commun. 2012, 425, 866–872.
- Beisswenger, P.; Ruggiero-Lopez, D. Metformin inhibition of glycation processes. Diabetes Metab. 2003, 29, 6S95–6S103.
- Yan, S.F.; D’Agati, V.; Schmidt, A.M.; Ramasamy, R. Receptor for Advanced Glycation Endproducts (RAGE): A formidable force in the pathogenesis of the cardiovascular complications of diabetes & aging. Curr. Mol. Med. 2007, 7, 699–710.
- Di Marco, E.; Gray, S.P.; Jandeleit-Dahm, K. Diabetes alters activation and repression of pro- and anti-inflammatory signaling pathways in the vasculature. Front. Endocrinol. 2013, 4, 68.
- Mamputu, J.C.; Wiernsperger, N.F.; Renier, G. Antiatherogenic properties of metformin: The experimental evidence. Diabetes Metab. 2003, 29, 6S71–6S76.
- Saisho, Y. Metformin and inflammation: Its potential beyond glucose-lowering effect. Endocr. Metab. Immune Disord. Drug Targets 2015, 15, 196–205.
- Ouslimani, N.; Peynet, J.; Bonnefont-Rousselot, D.; Thérond, P.; Legrand, A.; Beaudeux, J.L. Metformin decreases intracellular production of reactive oxygen species in aortic endothelial cells. Metabolism 2005, 54, 829–834.
- Bakhashab, S.; Ahmed, F.W.; Schulten, H.J.; Bashir, A.; Karim, S.; Al-Malki, A.L.; Gari, M.A.; Abuzenadah, A.M.; Chaudhary, A.G.; Alqahtani, M.H.; et al. Metformin improves the angiogenic potential of human CD34⁺ cells co-incident with downregulating CXCL10 and TIMP1 gene expression and increasing VEGFA under hyperglycemia and hypoxia within a therapeutic window for myocardial infarction. Cardiovasc. Diabetol. 2016, 15, 27.
- Yu, J.W.; Deng, Y.P.; Han, X.; Ren, G.F.; Cai, J.; Jiang, G.J. Metformin improves the angiogenic functions of endothelial progenitor cells via activating AMPK/eNOS pathway in diabetic mice. Cardiovasc. Diabetol. 2016, 15, 88.
- Arunachalam, G.; Samuel, S.M.; Marei, I.; Ding, H.; Triggle, C.R. Metformin modulates hyperglycaemia-induced endothelial senescence and apoptosis through SIRT1. Br. J. Pharmacol. 2014, 171, 523–535.
- Kinaan, M.; Ding, H.; Triggle, C.R. Metformin: An old drug for the treatment of diabetes but a new drug for the protection of the endothelium. Med. Princ. Pract. 2015, 24, 401–415.
- Eskens, B.J.; Zuurbier, C.J.; van Haare, J.; Vink, H.; van Teeffelen, J.W. Effects of two weeks of metformin treatment on whole-body glycocalyx barrier properties in db/db mice. Cardiovasc. Diabetol. 2013, 12, 175.
- Vega, M.; Mauro, M.; Williams, Z. Direct toxicity of insulin on the human placenta and protection by metformin. Fertil. Steril. 2019, 111, 489–496.e5.
- Tenório, M.B.; Ferreira, R.C.; Moura, F.A.; Bueno, N.B.; de Oliveira, A.C.M.; Goulart, M.O.F. Cross-talk between oxidative stress and inflammation in preeclampsia. Oxid. Med. Cell Longev. 2019, 2019, 8238727.
- Han, C.S.; Herrin, M.A.; Pitruzzello, M.C.; Mulla, M.J.; Werner, E.F.; Pettker, C.M.; Flannery, C.A.; Abrahams, V.M. Glucose and metformin modulate human first trimester trophoblast function: A model and potential therapy for diabetes-associated uteroplacental insufficiency. Am. J. Reprod. Immunol. 2015, 73, 362–371.
- Chiswick, C.; Reynolds, R.M.; Denison, F.; Drake, A.J.; Forbes, S.; Newby, D.E.; Walker, B.R.; Quenby, S.; Wray, S.; Weeks, A.; et al. Effect of metformin on maternal and fetal outcomes in obese pregnant women (EMPOWaR): A randomised, double-blind, placebo-controlled trial. Lancet Diabetes Endocrinol. 2015, 3, 778–786.
- Hu, J.; Zhang, J.; Zhu, B. Protective effect of metformin on a rat model of lipopolysaccharide-induced preeclampsia. Fundam. Clin. Pharmacol. 2019, 33, 649–658.
- Shu, C.; Yan, D.; Chen, C.; Mo, Y.; Wu, L.; Gu, J.; Shah, N.K.; He, J.; Dong, S. Metformin exhibits its therapeutic effect in the treatment of pre-eclampsia via modulating the Met/H19/miR-148a-5p/P28 and Met/H19/miR-216-3p/EBI3 signaling pathways. Int. Immunopharmacol. 2019, 74, 105693.
- Correia-Branco, A.; Keating, E.; Martel, F. Involvement of mTOR, JNK and PI3K in the negative effect of ethanol and metformin on the human first-trimester extravillous trophoblast HTR-8/SVneo cell line. Eur. J. Pharmacol. 2018, 833, 16–24.
- Triggle, C.R.; Ding, H. Metformin is not just an antihyperglycaemic drug but also has protective effects on the vascular endothelium. Acta Physiol. 2017, 219, 138–151.
- Austgulen, R.; Lien, E.; Vince, G.; Redman, C.W. Increased maternal plasma levels of soluble adhesion molecules (ICAM-1, VCAM-1, E-selectin) in preeclampsia. Eur. J. Obstet. Gynecol. Reprod. Biol. 1997, 71, 53–58.
- Chaiworapongsa, T.; Romero, R.; Yoshimatsu, J.; Espinoza, J.; Kim, Y.M.; Park, K.; Kalache, K.; Edwin, S.; Bujold, E.; Gomez, R. Soluble adhesion molecule profile in normal pregnancy and pre-eclampsia. J. Matern. Fetal. Neonatal. Med. 2002, 12, 19–27.
- Brownfoot, F.C.; Hastie, R.; Hannan, N.J.; Cannon, P.; Tuohey, L.; Parry, L.J.; Senadheera, S.; Illanes, S.E.; Kaitu’u-Lino, T.J.; Tong, S. Metformin as a prevention and treatment for preeclampsia: Effects on soluble fms-like tyrosine kinase 1 and soluble endoglin secretion and endothelial dysfunction. Am. J. Obstet. Gynecol. 2016, 214, 356.e1–356.e15.
- Kaitu’u-Lino, T.J.; Brownfoot, F.C.; Beard, S.; Cannon, P.; Hastie, R.; Nguyen, T.V.; Binder, N.K.; Tong, S.; Hannan, N.J. Combining metformin and esomeprazole is additive in reducing sFlt-1 secretion and decreasing endothelial dysfunction-implications for treating preeclampsia. PLoS ONE 2018, 13, e0188845.
- Soobryan, N.; Murugesan, S.; Pandiyan, A.; Moodley, J.; Mackraj, I. Angiogenic dysregulation in pregnancy-related hypertension-A role for metformin. Reprod. Sci. 2018, 25, 1531–1539.
- Brownfoot, F.C.; Hastie, R.; Hannan, N.J.; Cannon, P.; Nguyen, T.V.; Tuohey, L.; Cluver, C.; Tong, S.; Kaitu’u-Lino, T.J. Combining metformin and sulfasalazine additively reduces the secretion of antiangiogenic factors from the placenta: Implications for the treatment of preeclampsia. Placenta 2020, 95, 78–83.
- Brouillet, S.; Hoffmann, P.; Feige, J.J.; Alfaidy, N. EG-VEGF: A key endocrine factor in placental development. Trends Endocrinol. Metab. 2012, 23, 501–508.
- Jena, M.K.; Sharma, N.R.; Petitt, M.; Maulik, D.; Nayak, N.R. Pathogenesis of preeclampsia and therapeutic approaches targeting the placenta. Biomolecules 2020, 10, 953.
- Wang, F.; Cao, G.; Yi, W.; Li, L.; Cao, X. Effect of metformin on a preeclampsia-like mouse model induced by high-fat diet. Biomed. Res. Int. 2019, 2019, 6547019.
- Sutton, E.F.; Gemmel, M.; Powers, R.W. Nitric oxide signaling in pregnancy and preeclampsia. Nitric. Oxide. 2020, 95, 55–62.
- Liao, Y.F.; Chen, L.L.; Zeng, T.S.; Li, Y.M.; Fan, Y.U.; Hu, L.J.; Ling, Y. Number of circulating endothelial progenitor cells as a marker of vascular endothelial function for type 2 diabetes. Vasc. Med. 2010, 15, 279–285.
- Asadian, S.; Alibabrdel, M.; Daei, N.; Cheraghi, H.; Maedeh Jafari, S.; Noshadirad, E.; Jabarpour, M.; Siavashi, V.; Nassiri, S.M. Improved angiogenic activity of endothelial progenitor cell in diabetic patients treated with insulin plus metformin. J. Cell Biochem. 2018.
- Ni, H.Z.; Liu, Z.; Sun, L.L.; Zhou, M.; Liu, C.; Li, W.D.; Li, X.Q. Metformin inhibits angiogenesis of endothelial progenitor cells via miR-221-mediated p27 expression and autophagy. Future Med. Chem. 2019, 11, 2263–2272.
- Li, W.D.; Li, N.P.; Song, D.D.; Rong, J.J.; Qian, A.M.; Li, X.Q. Metformin inhibits endothelial progenitor cell migration by decreasing matrix metalloproteinases, MMP-2 and MMP-9, via the AMPK/mTOR/autophagy pathway. Int. J. Mol. Med. 2017, 39, 1262–1268.
- Xu, C.; Bailly-Maitre, B.; Reed, J.C. Endoplasmic reticulum stress: Cell life and death decisions. J. Clin. Investig. 2005, 115, 2656–2664.
- Burton, G.J.; Yung, H.W.; Cindrova-Davies, T.; Charnock-Jones, D.S. Placental endoplasmic reticulum stress and oxidative stress in the pathophysiology of unexplained intrauterine growth restriction and early onset preeclampsia. Placenta 2009, 30, S43–S48.
- Burton, G.J.; Yung, H.W.; Murray, A.J. Mitochondrial-Endoplasmic reticulum interactions in the trophoblast: Stress and senescence. Placenta 2017, 52, 146–155.
- Suzuki, M.; Mizuuchi, M.; Baba, T.; Fujibe, Y.; Ino, Y.; Ishioka, S.; Saito, T. The roles of metformin and pravastatin on placental endoplasmic reticulum stress and placental growth factor in human villous-Like trophoblastic BeWo cells. Sapporo Med. J. 2018, 87, 75–84.
- Isaka, K.; Usuda, S.; Ito, H.; Sagawa, Y.; Nakamura, H.; Nishi, H.; Suzuki, Y.; Li, Y.F.; Takayama, M. Expression and activity of matrix metalloproteinase 2 and 9 in human trophoblasts. Placenta 2003, 24, 53–64.
- Rajendrasozhan, S.; Yang, S.R.; Kinnula, V.L.; Rahman, I. SIRT1, an antiinflammatory and antiaging protein, is decreased in lungs of patients with chronic obstructive pulmonary disease. Am. J. Respir. Crit. Care Med. 2008, 177, 861–870.
- Brink, H.S.; Alkemade, M.; van der Lely, A.J.; van der Linden, J. Metformin in women at high risk of gestational diabetes mellitus. Diabetes Metab. 2018, 44, 300–302.
- Ainuddin, J.; Karim, N.; Hasan, A.A.; Naqvi, S.A. Metformin versus insulin treatment in gestational diabetes in pregnancy in a developing country: A randomized control trial. Diabetes Res. Clin. Pract. 2015, 107, 290–299.
- Tertti, K.; Ekblad, U.; Koskinen, P.; Vahlberg, T.; Rönnemaa, T. Metformin vs. insulin in gestational diabetes. A randomized study characterizing metformin patients needing additional insulin. Diabetes Obes. Metab. 2013, 15, 246–251.
- Niromanesh, S.; Alavi, A.; Sharbaf, F.R.; Amjadi, N.; Moosavi, S.; Akbari, S. Metformin compared with insulin in the management of gestational diabetes mellitus: A randomized clinical trial. Diabetes Res. Clin. Pract. 2012, 98, 422–429.
- Feig, D.S.; Donovan, L.E.; Zinman, B.; Sanchez, J.J.; Asztalos, E.; Ryan, E.A.; Fantus, I.G.; Hutton, E.; Armson, A.B.; Lipscombe, L.L.; et al. Metformin in women with type 2 diabetes in pregnancy (MiTy): A multicentre, international, randomised, placebo-controlled trial. Lancet Diabetes Endocrinol. 2020, 8, 834–844.
- Ainuddin, J.A.; Karim, N.; Zaheer, S.; Ali, S.S.; Hasan, A.A. Metformin treatment in type 2 diabetes in pregnancy: An active controlled, parallel-group, randomized, open label study in patients with type 2 diabetes in pregnancy. J. Diabetes Res. 2015, 2015, 325851.
- Nascimento, I.B.D.; Sales, W.B.; Dienstmann, G.; Souza, M.L.R.; Fleig, R.; Silva, J.C. Metformin for prevention of cesarean delivery and large-for-gestational-age newborns in non-diabetic obese pregnant women: A randomized clinical trial. Arch. Endocrinol. Metab. 2020, 64, 290–297.
- Syngelaki, A.; Nicolaides, K.H.; Balani, J.; Hyer, S.; Akolekar, R.; Kotecha, R.; Pastides, A.; Shehata, H. Metformin versus placebo in obese pregnant women without diabetes mellitus. N. Engl. J. Med. 2016, 374, 434–443.
- Løvvik, T.S.; Carlsen, S.M.; Salvesen, Ø.; Steffensen, B.; Bixo, M.; Gómez-Real, F.; Lønnebotn, M.; Hestvold, K.V.; Zabielska, R.; Hirschberg, A.L.; et al. Use of metformin to treat pregnant women with polycystic ovary syndrome (PregMet2): A randomised, double-blind, placebo-controlled trial. Lancet Diabetes Endocrinol. 2019, 7, 256–266.
- Prejbisz, A.; Dobrowolski, P.; Kosiński, P.; Bomba-Opoń, D.; Adamczak, M.; Bekiesińska-Figatowska, M.; Kądziela, J.; Konopka, A.; Kostka-Jeziorny, K.; Kurnatowska, I.; et al. Management of hypertension in pregnancy: Prevention, diagnosis, treatment and long-term prognosis. Kardiol. Pol. 2019, 77, 757–806.
- Brown, M.A.; Magee, L.A.; Kenny, L.C.; Karumanchi, S.A.; McCarthy, F.P.; Saito, S.; Hall, D.R.; Warren, C.E.; Adoyi, G.; Ishaku, S.; et al. International Society for the Study of Hypertension in Pregnancy (ISSHP). The hypertensive disorders of pregnancy: ISSHP classification, diagnosis & management recommendations for international practice. Pregnancy Hypertens. 2018, 13, 291–310.
- Magee, L.A.; Pels, A.; Helewa, M.; Rey, E.; von Dadelszen, P.; SOGC Hypertension Guideline Committee. Diagnosis, evaluation, and management of the hypertensive disorders of pregnancy: Executive summary. J. Obstet. Gynaecol. Can. 2014, 36, 575–576.
- Nascimento, I.B.D.; Dienstmann, G.; de Souza, M.L.R.; Fleig, R.; Hoffmann, C.B.P.C.; Silva, J.C. Evaluation of preeclampsia results after use of metformin in gestation: Systematic review and meta-analysis. Rev. Bras. Ginecol. Obstet. 2018, 40, 713–721.
- Kalafat, E.; Sukur, Y.E.; Abdi, A.; Thilaganathan, B.; Khalil, A. Metformin for prevention of hypertensive disorders of pregnancy in women with gestational diabetes or obesity: Systematic review and meta-analysis of randomized trials. Ultrasound Obstet. Gynecol. 2018, 52, 706–714.
- Butalia, S.; Gutierrez, L.; Lodha, A.; Aitken, E.; Zakariasen, A.; Donovan, L. Short- and long-term outcomes of metformin compared with insulin alone in pregnancy: A systematic review and meta-analysis. Diabet. Med. 2017, 34, 27–36.
- Feng, Y.; Yang, H. Metformin-A potentially effective drug for gestational diabetes mellitus: A systematic review and meta-analysis. J. Matern. Fetal. Neonatal. Med. 2017, 30, 1874–1881.
- Li, G.; Zhao, S.; Cui, S.; Li, L.; Xu, Y.; Li, Y. Effect comparison of metformin with insulin treatment for gestational diabetes: A meta-analysis based on RCTs. Arch. Gynecol. Obstet. 2015, 292, 111–120.
- Poolsup, N.; Suksomboon, N.; Amin, M. Efficacy and safety of oral antidiabetic drugs in comparison to insulin in treating gestational diabetes mellitus: A meta-analysis. PLoS ONE 2014, 9, e109985.
- Zhu, B.; Zhang, L.; Fan, Y.Y.; Wang, L.; Li, X.G.; Liu, T.; Cao, Y.S.; Zhao, Z.G. Metformin versus insulin in gestational diabetes mellitus: A meta-analysis of randomized clinical trials. Ir. J. Med. Sci. 2016, 185, 371–381.
- Gui, J.; Liu, Q.; Feng, L. Metformin vs insulin in the management of gestational diabetes: A meta-analysis. PLoS ONE 2013, 8, e64585.
- Dodd, J.M.; Grivell, R.M.; Deussen, A.R.; Hague, W.M. Metformin for women who are overweight or obese during pregnancy for improving maternal and infant outcomes. Cochrane Database Syst. Rev. 2018, 7, CD010564.
- Feng, L.; Lin, X.F.; Wan, Z.H.; Hu, D.; Du, Y.K. Efficacy of metformin on pregnancy complications in women with polycystic ovary syndrome: A meta-analysis. Gynecol. Endocrinol. 2015, 31, 833–839.
- Zheng, J.; Shan, P.F.; Gu, W. The efficacy of metformin in pregnant women with polycystic ovary syndrome: A meta-analysis of clinical trials. J. Endocrinol. Investig. 2013, 36, 797–802.