 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Levent M. Akyürek | + 1493 word(s) | 1493 | 2021-06-21 05:00:47 | | | |
| 2 | Bruce Ren | -20 word(s) | 1473 | 2021-07-06 04:03:57 | | | | |
| 3 | Bruce Ren | -20 word(s) | 1473 | 2021-07-06 04:04:14 | | |
Video Upload Options
Filamin A (FLNA) is a large actin-binding cytoskeletal protein that is important for cell motility by stabilizing actin networks and integrating them with cell membranes. Interestingly, a C-terminal fragment of FLNA can be cleaved off by calpain to stimulate adaptive angiogenesis by transporting multiple transcription factors into the nucleus. Recently, increasing evidence suggests that FLNA participates in the pathogenesis of cardiovascular and respiratory diseases, in which the interaction of FLNA with transcription factors and/or cell signaling molecules dictate the function of vascular cells. Localized FLNA mutations associate with cardiovascular malformations in humans. A lack of FLNA in experimental animal models disrupts cell migration during embryogenesis and causes anomalies, including heart and vessels, similar to human malformations.
1. Introduction
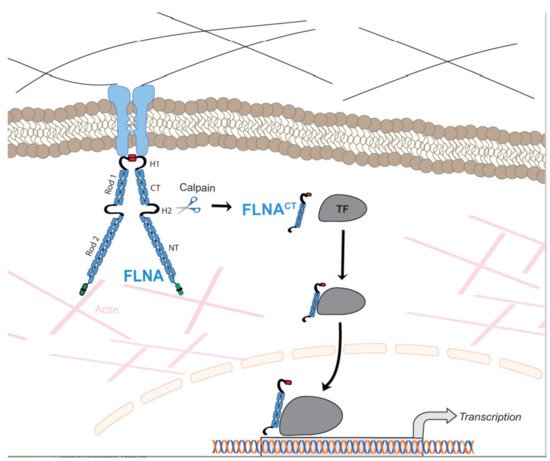
1.1. Filamin A in Cellular Signaling and Migration
1.2. Therapeutic Use of Calpain Inhibitors
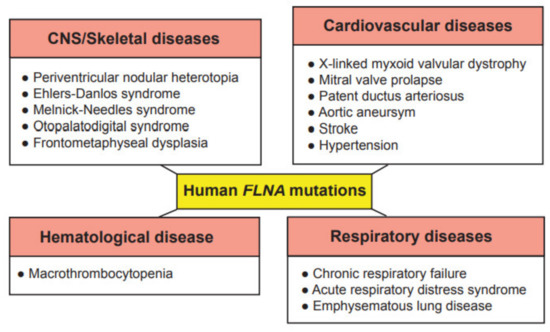
2. Genetic Disorders Associated with Human Filamin A Mutations
References
- Zhou, X.; Borén, J.; Akyürek, L.M. Filamins in cardiovascular development. Trends Cardiovasc. Med. 2007, 17, 222–229.
- Van der Flier, A.; Sonnenberg, A. Structural and functional aspects of filamins. Biochim. Biophys. Acta 2001, 1538, 99–117.
- Lu, J.; Lian, G.; Lenkinski, R.; De Grand, A.; Vaid, R.R.; Bryce, T.; Stasenko, M.; Boskey, A.; Walsh, C.; Sheen, V. Filamin B mutations cause chondrocyte defects in skeletal development. Hum. Mol. Genet. 2007, 16, 1661–1675.
- Stossel, T.P.; Condeelis, J.; Cooley, L.; Hartwig, J.H.; Noegel, A.; Schleicher, M.; Shapiro, S.S. Filamins as integrators of cell mechanics and signalling. Nat. Rev. Mol. Cell Biol. 2001, 2, 138–145.
- Zhou, A.-X.; Hartwig, J.H.; Akyürek, L.M. Filamins in cell signaling, transcription and organ development. Trends Cell Biol. 2010, 20, 113–123.
- Fox, J.E.; Goll, D.E.; Reynolds, C.C.; Phillips, D.R. Identification of two proteins (actin-binding protein and P235) that are hydrolyzed by endogenous Ca2+-dependent protease during platelet aggregation. J. Biol. Chem. 1985, 260, 1060–1066.
- Zhou, J.; Kang, X.; An, H.; Lv, Y.; Liu, X. The function and pathogenic mechanism of filamin A. Gene 2021, 784, 145575.
- Nakamura, F.; Stossel, T.P.; Hartwig, J.H. The filamins: Organizers of cell structure and function. Cell Adh. Migr. 2011, 5, 160–169.
- Campbell, I.D. Studies of focal adhesion assembly. Biochem. Soc. Trans. 2008, 36, 263–266.
- Bellanger, J.M.; Astier, C.; Sardet, C.; Ohta, Y.; Stossel, T.P.; Debant, A. The Rac1-and RhoG-specific GEF domain of Trio targets filamin to remodel cytoskeletal actin. Nat. Cell Biol. 2000, 2, 888–892.
- Urban, E.; Jacob, S.; Nemethova, M.; Resch, G.P.; Small, J.V. Electron tomography reveals unbranched networks of actin filaments in lamellipodia. Nat. Cell Biol. 2010, 12, 429–435.
- Nakamura, F.; Osborn, T.M.; Hartemink, C.A.; Hartwig, J.H.; Stossel, T.P. Structural basis of filamin A functions. J. Cell Biol. 2007, 179, 1011–1025.
- Urra, H.; Henriquez, D.R.; Cánovas, J.; Villarroel-Campos, D.; Carreras-Sureda, A.; Pulgar, E.; Molina, E.; Hazari, Y.M.; Limia, C.M.; Alvarez-Rojas, S.; et al. IRE1α governs cytoskeleton remodelling and cell migration through a direct interaction with filamin A. Nat. Cell Biol. 2018, 20, 942–953.
- Davies, P.; Wallach, D.; Willingham, M.; Pastan, I.; Yamaguchi, M.; Robson, R. Filamin-actin interaction. Dissociation of binding from gelation by Ca2+-activated proteolysis. J. Biol. Chem. 1978, 253, 4036–4042.
- Hemmings, L.; Rees, D.; Ohanian, V.; Bolton, S.; Gilmore, A.; Patel, B.; Priddle, H.; Trevithick, J.; Hynes, R.; Critchley, D. Talin contains three actin-binding sites each of which is adjacent to a vinculin-binding site. J. Cell Sci. 1996, 109, 2715–2726.
- Arthur, J.S.C.; Elce, J.S.; Hegadorn, C.; Williams, K.; Greer, P.A. Disruption of the murine calpain small subunit gene, Capn4: calpain is essential for embryonic development but not for cell growth and division. Mol. Cell. Biol. 2000, 20, 4474–4481.
- Kishimoto, A.; Mikawa, K.; Hashimoto, K.; Yasuda, I.; Tanaka, S.; Tominaga, M.; Kuroda, T.; Nishizuka, Y. Limited proteolysis of protein kinase C subspecies by calcium-dependent neutral protease (Calpain). J. Biol. Chem. 1989, 264, 4088–4092.
- Kidd, V.J.; Lahti, J.M.; Teitz, T. Proteolytic regulation of apoptosis. Semin. Cell Dev. Biol. 2000, 11, 191–201.
- Pariat, M.; Salvat, C.; Bebien, M.; Brockly, F.; Altieri, E.; Carillo, S.; Jariel-Encontre, I.; Piechaczyk, M. The sensitivity of c-Jun and c-Fos proteins to calpains depends on conformational determinants of the monomers and not on formation of dimers. Biochem. J. 2000, 345 Pt 1, 129–138.
- Watanabe, N.; Woude, G.F.V.; Ikawa, Y.; Sagata, N. Specific proteolysis of the c-mos proto-oncogene product by calpain on fertilization of Xenopus eggs. Nat. Cell Biol. 1989, 342, 505–511.
- Donkor, I. An updated patent review of calpain inhibitors (2012—2014). Expert Opin. Ther. Pat. 2014, 25, 17–31.
- Baghdiguian, S.; Martin, M.; Richard, I.; Pons, F.; Astier, C.; Bourg, N.; Hay, R.; Chemaly, R.; Halaby, G.; Loiselet, J.; et al. Calpain 3 deficiency is associated with myonuclear apoptosis and profound perturbation of the IκBα/NF-κB pathway in limb-girdle muscular dystrophy type 2A. Nat. Med. 1999, 5, 503–511.
- Ilian, M.A.; Gilmour, R.S.; Bickerstaffe, R. Quantification of ovine and bovine calpain I, calpain II, and calpastatin mRNA by ribonuclease protection assay. J. Anim. Sci. 1999, 77, 853–864.
- Inserte, J.; Hernando, V.; Garcia-Dorado, D. Contribution of calpains to myocardial ischaemia/reperfusion injury. Cardiovasc. Res. 2012, 96, 23–31.
- Leloup, L.; Wells, A. Calpains as potential anti-cancer targets. Expert Opin. Ther. Targets 2011, 15, 309–323.
- Fox, J.W.; Lamperti, E.D.; Ekşioğlu, Y.Z.; E Hong, S.; Feng, Y.; A Graham, D.; Scheffer, I.; Dobyns, W.B.; A Hirsch, B.; A Radtke, R.; et al. Mutations in filamin 1 Prevent Migration of Cerebral Cortical Neurons in Human Periventricular Heterotopia. Neuron 1998, 21, 1315–1325.
- Chen, M.H.; Choudhury, S.; Hirata, M.; Khalsa, S.K.; Chang, B.; Walsh, C.A. Thoracic aortic aneurysm in patients with loss of function Filamin A mutations: Clinical characterization, genetics, and recommendations. Am. J. Med. Genet. Part A 2018, 176, 337–350.
- Robertson, S.P.; Jenkins, Z.A.; Morgan, T.; Adès, L.; Aftimos, S.; Boute, O.; Fiskerstrand, T.; Garcia-Miñaur, S.; Grix, A.; Green, A.; et al. Frontometaphyseal dysplasia: Mutations in FLNA and phenotypic diversity. Am J. Med. Genet. 2006, 140A, 1726–1736.
- Robertson, S.P.; Twigg, S.R.F.; Sutherland-Smith, V.; Biancalana, V.; Gorlin, R.J.; Horn, D.; Kenwrick, S.J.; Kim, C.; Morava, E.; Newbury-Ecob, R.; et al. Localized mutations in the gene encoding the cytoskeletal protein filamin A cause diverse malformations in humans. Nat. Genet. 2003, 33, 487–491.
- Feng, Y.; Walsh, C.A. The many faces of filamin: A versatile molecular scaffold for cell motility and signalling. Nat. Cell Biol. 2004, 6, 1034–1038.
- Le Tourneau, T.; Le Scouarnec, S.; Cueff, C.; Bernstein, D.; Aalberts, J.J.; Lecointe, S.; Mérot, J.; Bernstein, J.A.; Oomen, T.; Dina, C.; et al. New insights into mitral valve dystrophy: A Filamin-A genotype–phenotype and outcome study. Eur. Hear. J. 2017, 39, 1269–1277.
- Kyndt, F.; Gueffet, J.-P.; Probst, V.; Jaafar, P.; Legendre, A.; Le Bouffant, F.; Toquet, C.; Roy, E.; McGregor, L.; Lynch, S.A.; et al. Mutations in the gene encoding filamin A as a cause for familial cardiac valvular dystrophy. Circulation 2007, 115, 40–49.
- Sasaki, A.; Masuda, Y.; Ohta, Y.; Ikeda, K.; Watanabe, K. Filamin associates with Smads and regulates transforming growth factor-β signaling. J. Biol. Chem. 2001, 276, 17871–17877.
- Li, X.; Lu, Y.; Wang, J.; Liu, M.; Wang, M.; Hu, L.; Du, W.; Wang, L.; Jiang, Z.; Gu, X.; et al. An integration-free iPSC line ZZUNEUi008-A derived from dermal fibroblasts of a child with cardiac valvular dysplasia carrying a mutation in FLNA gene. Stem Cell Res. 2020, 47, 101882.
- Parrini, E.; Ramazzotti, A.; Dobyns, W.B.; Mei, D.; Moro, F.; Veggiotti, P.; Marini, C.; Brilstra, E.H.; Bernardina, B.D.; Goodwin, L.; et al. Periventricular heterotopia: Phenotypic heterogeneity and correlation with filamin A mutations. Brain 2006, 129, 1892–1906.
- Sheen, V.L.; Jansen, A.; Chen, M.H.; Parrini, E.; Morgan, T.; Ravenscroft, R.; Ganesh, V.; Underwood, T.; Wiley, J.; Leventer, R.; et al. Filamin A mutations cause periventricular heterotopia with Ehlers-Danlos syndrome. Neurology 2005, 64, 254–262.
- Sasaki, E.; Byrne, A.T.; Phelan, E.; Cox, D.; Reardon, W. A review of filamin A mutations and associated interstitial lung disease. Eur. J. Nucl. Med. Mol. Imaging 2019, 178, 121–129.
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Committee ALQA; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424.


