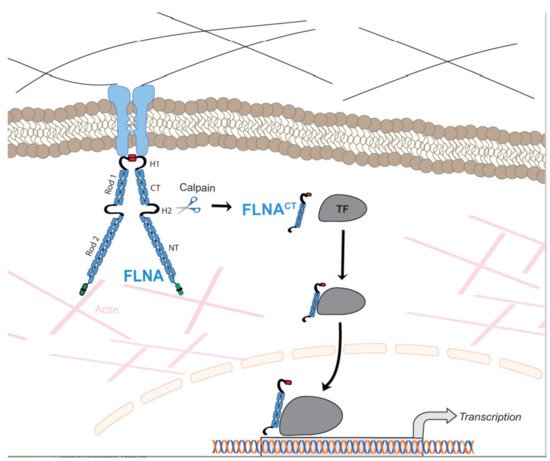
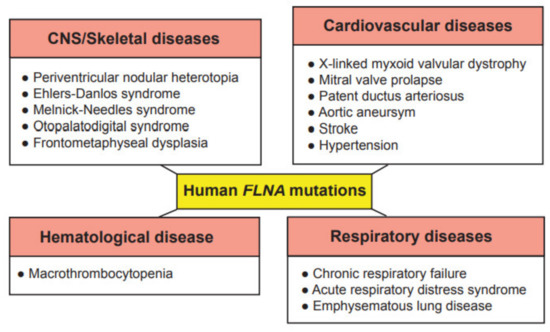
Due to the wide range of function of FLNA in cell migration and cell signaling, mutations in the
FLNA gene cause a varying spectrum of developmental malformations, which are also associated with cardiovascular malformations and diseases (), whereas
FLNB and
FLNC mutations are mainly restricted to skeletal and cardiac muscle diseases, respectively. As these proteins have gained interest recently in the research field, their more predominant functions are yet to be explored. The first mutation to be identified in the
FLNA gene was the null mutation, where coding amino acids are converted to a stop codon in exon 3, resulting in periventricular nodular heterotopia (PVNH), where a six-layer neocortex is not formed due to failed neuronal migration [
26]. PVNH is mainly linked to females, while the majority of hemizygous males are confined to embryonic lethality, and live-born males display aortic dilation and die from a massive hemorrhage in the neonatal period [
26]. Female PVNH patients run a high risk of strokes and are associated with other cardiovascular abnormalities, such as valvular abnormalities, persistent ductus arteriosus and aneurysms in the aorta [
26]. The majority of PVNH patients with
FLNA mutations also have thoracic aortic aneurysms [
27]. Multiple missense mutations, resulting in substitutions in the actin-binding domain (ABD), have been identified, mainly in the Ig repeats of 9, 10, 14, 16, 22 and 23 of FLNA [
28]. These mutations are found in otopalatodigital syndrome, Melnick-Needles syndrome and front metaphyseal dysplasia [
28], and are also accompanied by cardiac, tracheobronchial and urological malformations, resulting in perinatal death [
29]. However, phenotypes caused by the otopalatodigital syndrome, Melnick-Needles syndrome and front metaphyseal dysplasia are very distinct from PVNH [
30].
Missense mutations in the
N-terminal terminal region of the
FLNA gene are associated with non-syndromic mitral valve dystrophy [
31]. The genomic deletion of codons from exon 19 results in cardiac X-linked myxoid valvular dystrophy (XVMD), a specific cardiovascular malformation [
32]. XVMD are frequently involved in vascular anomalies, which include mitral valve prolapse, as well as mitral and aortic regurgitation. Interestingly, it is known that a defective signaling cascade in TGF-β results in impaired mitral valve remodeling, and FLNA contributes to changes in cardiac valves by regulating the TGF-β signaling cascade via interaction with SMADs [
33]. XVMD are not linked with PVNH and the other congenital disorders mentioned above. Interestingly, a human-induced pluripotent stem cell line from a 10-year-old male patient with cardiac valvular dysplasia, carrying a particular mutation in the
FLNA gene (c. 84G→A), using non-integrative Sendai virus reprogramming technology, has been established to the study molecular mechanisms of cardiac valvular dysplasia, as this cell line can differentiate into three germ layers in vivo [
34].
Exon 21 nonsense mutations, frameshift and 4-shift mutations have been identified in FLNA to cause bilateral PVNH, along with Ehlers-Danlos syndrome, accompanied by minor cardiovascular malformations [
35]. A variant of PVNH, associated with
FLNA mutations and Ehlers-Danlos syndrome, leads to the development of aortic dilation during early childhood [
36]. Novel pathogenic variants have been identified in the
FLNA gene, causing respiratory failure in newborns [
37]. In summary, these human
FLNA mutations are classified as at least moderate pathogenic associations according to the guidelines provided by the American College of Medical Genetics and Genomics [
38].