 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Eve-Isabelle PECHEUR | + 21261 word(s) | 21261 | 2021-05-27 06:05:36 | | | |
| 2 | Eve-Isabelle PECHEUR | -8577 word(s) | 12684 | 2021-05-28 03:34:58 | | | | |
| 3 | Peter Tang | -7353 word(s) | 5331 | 2021-05-28 09:42:57 | | | | |
| 4 | Peter Tang | Meta information modification | 5331 | 2021-06-01 05:28:34 | | | | |
| 5 | Conner Chen | Meta information modification | 5331 | 2021-09-22 04:07:09 | | |
Video Upload Options
Chronic infection by the hepatitis C virus (HCV) is a major cause of liver diseases, predisposing to fibrosis and hepatocellular carcinoma. Liver fibrosis is characterized by an overly abundant accumulation of components of the hepatic extracellular matrix, such as collagen and elastin, with consequences on the properties of this microenvironment and cancer initiation and growth. This review will provide an update on mechanistic concepts of HCV-related liver fibrosis/cirrhosis and early stages of carcinogenesis, with a dissection of the molecular details of the cross-talk during disease progression between hepatocytes, the extracellular matrix and hepatic stellate cells.
1. Introduction
|
Virus |
HBV |
HCV |
|---|---|---|
|
Viral family |
Hepadnaviridae |
Flaviviridae |
|
Genome |
DNA and cccDNA |
RNA |
|
Life cycle |
Genome integration, expression of HBx protein, insertional activation of cellular oncogenes, cccDNA (minichromosome) |
Exclusively cytoplasmic |
|
Persistence |
Nucleus-located cccDNA |
Chronic inflammation, oxidative stress, alterations in cellular signaling and metabolism |
2. Main Actors of Liver Fibrosis
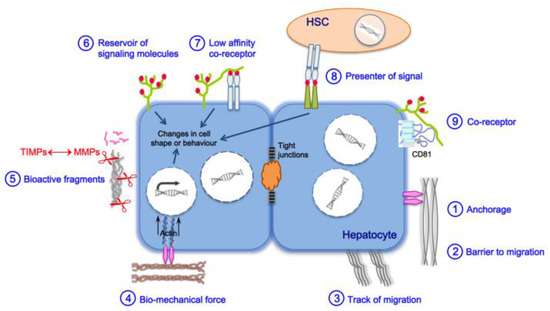
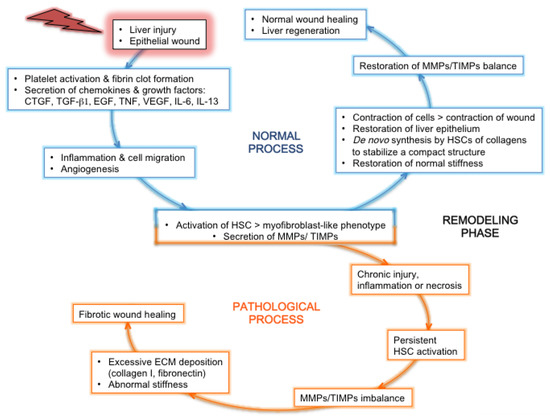
3. Liver Fibrosis and Cirrhosis
3.1. General Pan-Etiology Features
3.2. Features Linked to HCV Pathogenesis
|
HCV Proteins |
ECM Proteins or Cytokines |
|---|---|
|
Capsid core |
LOX ∞ [61] Procollagen I ∞ [62] Collagen I ∞ [61] MMP-2 ∞ [58] |
|
MMP-9 ∞ [63] |
|
|
COX-2 ∞ [63] |
|
|
Syndecan-1 * [31] |
|
|
Thrombospondin-1 ∞ [61] |
|
|
CTGF ∞ [58] |
|
|
TGF-β2 ◊ [67] |
|
|
Endoglin ∞ [68] |
|
|
Envelope glycoproteins E1 and/or E2 |
Glypican-3 * [69] |
|
TGF-β1 ◊ [66] |
|
|
Cysteine autoprotease NS2 |
MICA ∞ [70] TGF-β2 ◊ [67] |
|
Serine protease and helicase NS3 |
Procollagen I ∞ [62] MMP-9 ∞ [71] |
|
COX-2 ∞ [71] |
|
|
Thrombospondin-1 [72] |
|
|
Osteopontin * [64] |
|
|
TGF-β type I receptor * [73] |
|
|
NS3 with its cofactor NS4A |
MMP-9 ∞ [71] |
|
COX-2 ∞ [71] MICA ∞ [74] |
|
|
NS4B |
MMP-2 ∞ [76] |
|
NS5A |
MMP-2 ∞ [63] |
|
MMP-9 ∞ [63] |
|
|
COX-2 ∞ [63] |
|
|
Thrombospondin-1 ∞ [72] |
|
|
Osteopontin * [64] |
|
|
RNA-dependent RNA polymerase NS5B |
Osteopontin * [64] |
|
MICA ∞ [70] TGF-β ◊ [75] |
|
ECM Proteins/Cytokine |
F0/F1 |
F2 |
F3 |
F4 |
HCC |
References |
|---|---|---|---|---|---|---|
|
Collagens I, III, V |
F1 |
|||||
|
Collagen XII |
||||||
|
Collagen XIV |
||||||
|
Collagen XVI |
[59] |
|||||
|
Collagen XVIII |
[59] |
|||||
|
PIIINP |
F1 |
|||||
|
MMP-2, -7, -9 |
F1 |
|||||
|
TIMP-1 |
||||||
|
ADAM-TS1 |
[93] |
|||||
|
ADAM-TS2 |
[94] |
|||||
|
Xylosyltransferase-2 |
F1 |
|||||
|
Glypican-3 |
||||||
|
Hyaluronic acid |
||||||
|
Decorin |
F1 |
[92] |
||||
|
Biglycan |
[59] |
|||||
|
Fibromodulin |
[60] |
|||||
|
Lumican |
||||||
|
Versican |
F1 |
|||||
|
Tenascin-C |
||||||
|
Osteopontin |
F1 |
|||||
|
Fibronectin |
||||||
|
Fibronectin isoforms |
[108] |
|||||
|
Elastin |
||||||
|
MFAP-4 † |
F1 |
|||||
|
Fibulin-5 |
[84] |
|||||
|
TGF-β1 (protein, mRNA) |
||||||
|
TGF-β1 (serum levels) |
F1 |
|||||
|
TGF-β2 |
F1 |
F0 |
[67] |
|||
|
Endoglin (protein, serum levels) |
[100] |
|||||
|
Endoglin (mRNA) § |
[68] |
a Color codes: green, upregulation; dark green: higher upregulation; blue, downregulation; dark blue: higher downregulation; grey, no change; magenta, no correlation with liver fibrosis stage. † MFAP-4, microfibrillar-associated protein-4 (associated with elastin fibers). § Endoglin mRNA was found upregulated in chronically HCV-infected patients compared to noninfected patients but not correlating with liver fibrosis stage.
4. Are HSCs Direct Targets of HCV Infection?
5. Fibrosis Reversal in the Era of DAAs in HCV-Induced Liver Fibrosis
6. Conclusions and Perspectives
References
- Plummer, M.; de Martel, C.; Vignat, J.; Ferlay, J.; Bray, F.; Franceschi, S. Global burden of cancers attributable to infections in 2012: A synthetic analysis. Lancet Glob. Heal. 2016, 4, e609–e616.
- Villanueva, A. Hepatocellular Carcinoma. N. Engl. J. Med. 2019, 380, 1450–1462.
- Mitchell, J.K.; Lemon, S.M.; McGivern, D.R. How do persistent infections with hepatitis C virus cause liver cancer? Curr. Opin. Virol. 2015, 14, 101–108.
- Lemon, S.M.; McGivern, D.R. Is Hepatitis C Virus Carcinogenic? Gastroenterology 2012, 142, 1274–1278.
- World Health Organization. Combating Hepatitis B and C to Reach Elimination by 2030; World Health Organization: Geneva, Switzerland, 2016.
- Blach, S.; Zeuzem, S.; Manns, M.; Altraif, I.; Duberg, A.-S.; Muljono, D.H.; Waked, I.; Alavian, S.M.; Lee, M.-H.; Negro, F.; et al. Global prevalence and genotype distribution of hepatitis C virus infection in 2015: A modelling study. Lancet Gastroenterol. Hepatol. 2017, 2, 161–176.
- World Health Organization. Hepatitis C. Available online: (accessed on 29 January 2018).
- Hajarizadeh, B.; Grebely, J.; Dore, G.J. Epidemiology and natural history of HCV infection. Nat. Rev. Gastroenterol. Hepatol. 2013, 10, 553–562.
- Nault, J.-C.; Colombo, M. Hepatocellular carcinoma and direct acting antiviral treatments: Controversy after the revolution. J. Hepatol. 2016, 65, 663–665.
- Baumert, T.F.; Jühling, F.; Ono, A.; Hoshida, Y. Hepatitis C-related hepatocellular carcinoma in the era of new generation antivirals. BMC Med. 2017, 15, 1–10.
- Hamdane, N.; Jühling, F.; Crouchet, E.; Saghire, H.E.; Thumann, C.; Oudot, M.A.; Bandiera, S.; Saviano, A.; Ponsolles, C.; Suarez, A.A.R.; et al. HCV-Induced Epigenetic Changes Associated with Liver Cancer Risk Persist After Sustained Virologic Response. Gastroenterology 2019, 156, 2313–2329.
- Hengst, J.; Falk, C.S.; Schlaphoff, V.; Deterding, K.; Manns, M.P.; Cornberg, M.; Wedemeyer, H. Direct-Acting Antiviral-Induced Hepatitis C Virus Clearance Does Not Completely Restore the Altered Cytokine and Chemokine Milieu in Patients with Chronic Hepatitis C. J. Infect. Dis. 2016, 214, 1965–1974.
- Akuta, N.; Suzuki, F.; Hirakawa, M.; Kawamura, Y.; Sezaki, H.; Suzuki, Y.; Hosaka, T.; Kobayashi, M.; Kobayashi, M.; Saitoh, S.; et al. Amino acid substitutions in hepatitis C virus core region predict hepatocarcinogenesis following eradication of HCV RNA by antiviral therapy. J. Med. Virol. 2011, 83, 1016–1022.
- Takeda, H.; Takai, A.; Iguchi, E.; Mishima, M.; Arasawa, S.; Kumagai, K.; Eso, Y.; Shimizu, T.; Takahashi, K.; Ueda, Y.; et al. Oncogenic transcriptomic profile is sustained in the liver after the eradication of the hepatitis C virus. Carcinogenesis 2021.
- Paul, D.; Madan, V.; Bartenschlager, R. Hepatitis C Virus RNA Replication and Assembly: Living on the Fat of the Land. Cell Host Microbe 2014, 16, 569–579.
- Sebae, G.K.E.; Malatos, J.M.; Cone, M.-K.E.; Rhee, S.; Angelo, J.R.; Mager, J.; Tremblay, K.D. Single-cell murine genetic fate mapping reveals bipotential hepatoblasts and novel multi-organ endoderm progenitors. Development 2018, 145.
- Lee, Y.A.; Wallace, M.C.; Friedman, S.L. Pathobiology of liver fibrosis: A translational success story. Gut 2015, 64, 830–841.
- Rauterberg, J.; Voss, B.; Pott, G.; Gerlach, U. Connective tissue components of the normal and fibrotic liver. Klin. Wochenschr. 1981, 59, 767–779.
- Friedman, S.L. Hepatic Stellate Cells: Protean, Multifunctional, and Enigmatic Cells of the Liver. Physiol. Rev. 2008, 88, 125–172.
- De Minicis, S.; Seki, E.; Uchinami, H.; Kluwe, J.; Zhang, Y.; Brenner, D.A.; Schwabe, R.F. Gene Expression Profiles during Hepatic Stellate Cell Activation in Culture and in Vivo. Gastroenterology 2007, 132, 1937–1946.
- Lin, X.Z.; Horng, M.H.; Sun, Y.N.; Shiesh, S.C.; Chow, N.H.; Guo, X.Z. Computer morphometry for quantitative measurement of liver fibrosis: Comparison with knodell’s score, colorimetry and conventional description reports. J. Gastroenterol. Hepatol. 1998, 13, 75–80.
- Duarte, S.; Baber, J.; Fujii, T.; Coito, A.J. Matrix metalloproteinases in liver injury, repair and fibrosis. Matrix Biol. 2015, 44–46, 147–156.
- Rojkind, M.; Giambrone, M.A.; Biempica, L. Collagen Types in Normal and Cirrhotic Liver. Gastroenterology 1979, 76, 710–719.
- Kagan, H.M. Lysyl Oxidase: Mechanism, Regulation and Relationship to Liver Fibrosis. Pathol. Res. Pr. 1994, 190, 910–919.
- Andez, A.M.; Amenta, P.S. The extracellular matrix in hepatic regeneration. FASEB J. 1995, 9, 1401–1410.
- Iozzo, R.V.; Schaefer, L. Proteoglycan form and function: A comprehensive nomenclature of proteoglycans. Matrix Biol. 2015, 42, 11–55.
- Kanta, J. Elastin in the Liver. Front. Physiol. 2016, 7, 491.
- Musso, O.; Rehn, M.; Saarela, J.; Théret, N.; Liétard, J.; Hintikka, E.; Lotrian, D.; Campion, J.-P.; Pihlajaniemi, T.; Clément, B. Collagen XVIII is localized in sinusoids and basement membrane zones and expressed by hepatocytes and activated stellate cells in fibrotic human liver. Hepatology 1998, 28, 98–107.
- Ricard-Blum, S.; Vallet, S.D. Fragments generated upon extracellular matrix remodeling: Biological regulators and potential drugs. Matrix Biol. 2019, 75–76, 170–189.
- Sun, S.; Song, Z.; Cotler, S.J.; Cho, M. Biomechanics and functionality of hepatocytes in liver cirrhosis. J. Biomech. 2014, 47, 2205–2210.
- Grigorov, B.; Reungoat, E.; Maurin, A.G.D.; Varbanov, M.; Blaising, J.; Michelet, M.; Manuel, R.; Parent, R.; Bartosch, B.; Zoulim, F.; et al. Hepatitis C virus infection propagates through interactions between Syndecan-1 and CD81 and impacts the hepatocyte glycocalyx. Cell. Microbiol. 2017, 19.
- Wells, J.M.; Gaggar, A.; Blalock, J.E. MMP generated matrikines. Matrix Biol. 2015, 44–46, 122–129.
- Schuppan, D.; Ashfaq-Khan, M.; Yang, A.T.; Kim, Y.O. Liver fibrosis: Direct antifibrotic agents and targeted therapies. Matrix Biol. 2018, 68–69, 435–451.
- Ricard-Blum, S.; Baffet, G.; Théret, N. Molecular and tissue alterations of collagens in fibrosis. Matrix Biol. 2018.
- Jung, Y.; Witek, R.P.; Syn, W.-K.; Choi, S.S.; Omenetti, A.; Premont, R.; Guy, C.D.; Diehl, A.M. Signals from dying hepatocytes trigger growth of liver progenitors. Gut 2010, 59, 655–665.
- Wynn, T.A. Cellular and molecular mechanisms of fibrosis. J. Pathol 2008, 214, 199–210.
- Tsochatzis, E.A.; Bosch, J.; Burroughs, A.K. Liver cirrhosis. Lancet 2014, 383, 1749–1761.
- Zhao, S.-X.; Li, W.-C.; Fu, N.; Kong, L.-B.; Zhang, Q.-S.; Han, F.; Ren, W.-G.; Cui, P.; Du, J.-H.; Wang, B.-Y.; et al. CD14+ monocytes and CD163+ macrophages correlate with the severity of liver fibrosis in patients with chronic hepatitis C. Exp. Ther. Med. 2020, 20, 228.
- Douam, F.; Lavillette, D.; Cosset, F.-L. The Mechanism of HCV Entry into Host Cells. Prog. Mol. Biol. Transl. Sci. 2015, 129, 63–107.
- Hemler, M.E. Tetraspanin functions and associated microdomains. Nat. Rev. Mol. Cell Biol. 2005, 6, 801–811.
- Berditchevski, F. Complexes of tetraspanins with integrins: More than meets the eye. J. Cell Sci. 2001, 114, 4143–4151.
- Alisi, A.; Arciello, M.; Petrini, S.; Conti, B.; Missale, G.; Balsano, C. Focal Adhesion Kinase (FAK) Mediates the Induction of Pro-Oncogenic and Fibrogenic Phenotypes in Hepatitis C Virus (HCV)-Infected Cells. PLoS ONE 2012, 7, e44147.
- Vicente-Manzanares, M.; Webb, D.J.; Horwitz, A.R. Cell migration at a glance. J. Cell Sci. 2005, 118, 4917–4919.
- Martínez, S.M.; Crespo, G.; Navasa, M.; Forns, X. Noninvasive assessment of liver fibrosis. Hepatology 2011, 53, 325–335.
- Karsdal, M.A.; Manon-Jensen, T.; Genovese, F.; Kristensen, J.H.; Nielsen, M.J.; Sand, J.M.B.; Hansen, N.-U.B.; Bay-Jensen, A.-C.; Bager, C.L.; Krag, A.; et al. Novel insights into the function and dynamics of extracellular matrix in liver fibrosis. Am. J. Physiol. Liver Physiol. 2015, 308, G807–G830.
- Ferrell, L. Liver Pathology: Cirrhosis, Hepatitis, and Primary Liver Tumors—Update and Diagnostic Problems. Mod. Pathol. 2000, 13, 679–704.
- Govaere, O.; Cockell, S.; Van Haele, M.; Wouters, J.; Van Delm, W.; Van den Eynde, K.; Bianchi, A.; Van Eijsden, R.; Van Steenbergen, W.; Monbaliu, D.; et al. High-throughput sequencing identifies aetiology-dependent differences in ductular reaction in human chronic liver disease. J. Pathol. 2019, 248, 66–76.
- Trivedi, S.; Murthy, S.; Sharma, H.; Hartlage, A.S.; Kumar, A.; Gadi, S.; Simmonds, P.; Chauhan, L.V.; Scheel, T.K.H.; Billerbeck, E.; et al. Viral persistence, liver disease and host response in hepatitis c-like virus rat model. Hepatology 2017.
- Khatun, M.; Ray, R.B. Mechanisms Underlying Hepatitis C Virus-Associated Hepatic Fibrosis. Cells 2019, 8, 1249.
- Nielsen, M.J.; Karsdal, M.A.; Kazankov, K.; Grønbaek, H.; Krag, A.; Leeming, D.J.; Schuppan, D.; George, J. Fibrosis is not just fibrosis-basement membrane modelling and collagen metabolism differs between hepatitis B- and C-induced injury. Aliment. Pharmacol. Ther. 2016, 44, 1242–1252.
- Guido, M.; Mangia, A.; Faa, G. Chronic viral hepatitis: The histology report. Dig. Liver Dis. 2011, 43, S331–S343.
- Beltra, J.-C.; Decaluwe, H. Cytokines and persistent viral infections. Cytokine 2016, 82, 4–15.
- Govaere, O.; Petz, M.; Wouters, J.; Vandewynckel, Y.-P.; Scott, E.J.; Topal, B.; Nevens, F.; Verslype, C.; Anstee, Q.M.; Van Vlierberghe, H.; et al. The PDGFRα-laminin B1-keratin 19 cascade drives tumor progression at the invasive front of human hepatocellular carcinoma. Oncogene 2017, 36, 6605–6616.
- Martinez-Quetglas, I.; Pinyol, R.; Dauch, D.; Torrecilla, S.; Tovar, V.; Moeini, A.; Alsinet, C.; Portela, A.; Rodriguez-Carunchio, L.; Solé, M.; et al. IGF2 Is Up-Regulated by Epigenetic Mechanisms in Hepatocellular Carcinomas and is an Actionable Oncogene Product in Experimental Models. Gastroenterology 2016, 151, 1192–1205.
- Preisser, L.; Miot, C.; Le Guillou-Guillemette, H.; Beaumont, E.; Foucher, E.D.; Garo, E.; Blanchard, S.; Frémaux, I.; Croué, A.; Fouchard-Hubert, I.; et al. IL-34 and macrophage colony-stimulating factor are overexpressed in hepatitis C virus fibrosis and induce profibrotic macrophages that promote collagen synthesis by hepatic stellate cells. Hepatology 2014, 60, 1879–1890.
- Tummala, K.S.; Brandt, M.; Teijeiro, A.; Graña, O.; Schwabe, R.F.; Perna, C.; Djouder, N. Hepatocellular Carcinomas Originate Predominantly from Hepatocytes and Benign Lesions from Hepatic Progenitor Cells. Cell Rep. 2017, 19, 584–600.
- Schulze-Krebs, A.; Preimel, D.; Popov, Y.; Bartenschlager, R.; Lohmann, V.; Pinzani, M.; Schuppan, D. Hepatitis C Virus-Replicating Hepatocytes Induce Fibrogenic Activation of Hepatic Stellate Cells. Gastroenterology 2005, 129, 246–258.
- Shin, J.Y.; Hur, W.; Wang, J.S.; Jang, J.W.; Kim, C.W.; Bae, S.H.; Jang, S.K.; Yang, S.-H.; Sung, Y.C.; Kwon, O.-J.; et al. HCV core protein promotes liver fibrogenesis via up-regulation of CTGF with TGF-β1. Exp. Mol. Med. 2005, 37, 138–145.
- Baiocchini, A.; Montaldo, C.; Conigliaro, A.; Grimaldi, A.; Correani, V.; Mura, F.; Ciccosanti, F.; Rotiroti, N.; Brenna, A.; Montalbano, M.; et al. Extracellular Matrix Molecular Remodeling in Human Liver Fibrosis Evolution. PLoS ONE 2016, 11, e0151736.
- Mormone, E.; Lu, Y.; Ge, X.; Fiel, M.I.; Nieto, N. Fibromodulin, an Oxidative Stress-Sensitive Proteoglycan, Regulates the Fibrogenic Response to Liver Injury in Mice. Gastroenterology 2012, 142, 612–621.
- Benzoubir, N.; Lejamtel, C.; Battaglia, S.; Testoni, B.; Benassi, B.; Gondeau, C.; Perrin-Cocon, L.; Desterke, C.; Thiers, V.; Samuel, D.; et al. HCV core-mediated activation of latent TGF-β via thrombospondin drives the crosstalk between hepatocytes and stromal environment. J. Hepatol. 2013, 59, 1160–1168.
- Bataller, R.; Paik, Y.-H.; Lindquist, J.N.; Lemasters, J.J.; Brenner, D.A. Hepatitis C virus core and nonstructural proteins induce fibrogenic effects in hepatic stellate cells. Gastroenterology 2004, 126, 529–540.
- Nunez, O.; Fernández-Martínez, A.; Majano, P.L.; Apolinario, A.; Gómez-Gonzalo, M.; Benedicto, I.; López-Cabrera, M.; Boscá, L.; Clemente, G.; García-Monzón, C.; et al. Increased intrahepatic cyclooxygenase 2, matrix metalloproteinase 2, and matrix metalloproteinase 9 expression is associated with progressive liver disease in chronic hepatitis C virus infection: Role of viral core and NS5A proteins. Gut 2004, 53, 1665–1672.
- Iqbal, J.; Sarkar-Dutta, M.; McRae, S.; Ramachandran, A.; Kumar, B.; Waris, G. Osteopontin Regulates Hepatitis C Virus (HCV) Replication and Assembly by Interacting with HCV Proteins and Lipid Droplets and by Binding to Receptors AVβ3 and CD44. J. Virol. 2018, 92.
- Shirasaki, T.; Honda, M.; Yamashita, T.; Nio, K.; Shimakami, T.; Shimizu, R.; Nakasyo, S.; Murai, K.; Shirasaki, N.; Okada, H.; et al. The osteopontin-CD44 axis in hepatic cancer stem cells regulates IFN signaling and HCV replication. Sci. Rep. 2018, 8, 1–12.
- Jee, M.H.; Hong, K.Y.; Park, J.H.; Lee, J.S.; Kim, H.S.; Lee, S.H.; Jang, S.K. New Mechanism of Hepatic Fibrogenesis: Hepatitis C Virus Infection Induces Transforming Growth Factor Β1 Production through Glucose-Regulated Protein 94. J. Virol. 2015, 90, 3044–3055.
- Chida, T.; Ito, M.; Nakashima, K.; Kanegae, Y.; Aoshima, T.; Takabayashi, S.; Kawata, K.; Nakagawa, Y.; Yamamoto, M.; Shimano, H.; et al. Critical role of CREBH-mediated induction of transforming growth factor β2 by hepatitis C virus infection in fibrogenic responses in hepatic stellate cells. Hepatology 2017, 66, 1430–1443.
- Kwon, Y.-C.; Sasaki, R.; Meyer, K.; Ray, R. Hepatitis C Virus Core Protein Modulates Endoglin (CD105) Signaling Pathway for Liver Pathogenesis. J. Virol. 2017, 91.
- Xue, Y.; Mars, W.M.; Bowen, W.; Singhi, A.D.; Stoops, J.; Michalopoulos, G.K. Hepatitis C Virus Mimics Effects of Glypican-3 on CD81 and Promotes Development of Hepatocellular Carcinomas via Activation of Hippo Pathway in Hepatocytes. Am. J. Pathol. 2018, 188, 1469–1477.
- Kim, H.; Bose, S.K.; Meyer, K.; Ray, R.; Lyles, D.S. Hepatitis C Virus Impairs Natural Killer Cell-Mediated Augmentation of Complement Synthesis. J. Virol. 2014, 88, 2564–2571.
- Lu, L.; Zhang, Q.; Wu, K.; Chen, X.; Zheng, Y.; Zhu, C.; Wu, J. Hepatitis C virus NS3 protein enhances cancer cell invasion by activating matrix metalloproteinase-9 and cyclooxygenase-2 through ERK/p38/NF-κB signal cascade. Cancer Lett. 2015, 356, 470–478.
- Presser, L.D.; Haskett, A.; Waris, G. Hepatitis C virus-induced furin and thrombospondin-1 activate TGF-β1: Role of TGF-β1 in HCV replication. Virology 2011, 412, 284–296.
- Sakata, K.; Hara, M.; Terada, T.; Watanabe, N.; Takaya, D.; Yaguchi, S.-I.; Matsumoto, T.; Matsuura, T.; Shirouzu, M.; Yokoyama, S.; et al. HCV NS3 protease enhances liver fibrosis via binding to and activating TGF-β type I receptor. Sci. Rep. 2013, 3, 3243.
- Wen, C.; He, X.; Ma, H.; Hou, N.; Wei, C.; Song, T.; Zhang, Y.; Sun, L.; Ma, Q.; Zhong, H. Hepatitis C Virus Infection Downregulates the Ligands of the Activating Receptor NKG2D. Cell. Mol. Immunol. 2008, 5, 475–478.
- Verga-Gérard, A.; Porcherot, M.; Meyniel-Schicklin, L.; André, P.; Lotteau, V.; Perrin-Cocon, L. Hepatitis C virus/human interactome identifies SMURF2 and the viral protease as critical elements for the control of TGF-β signaling. FASEB J. 2013, 27, 4027–4040.
- Li, Y.; Zhang, Q.; Liu, Y.; Luo, Z.; Kang, L.; Qu, J.; Liu, W.; Xia, X.; Wu, K.; Wu, J. Hepatitis C Virus Activates Bcl-2 and MMP-2 Expression through Multiple Cellular Signaling Pathways. J. Virol. 2012, 86, 12531–12543.
- Chusri, P.; Kumthip, K.; Hong, J.; Zhu, C.; Duan, X.; Jilg, N.; Fusco, D.N.; Brisac, C.; Schaefer, E.A.; Cai, D.; et al. HCV induces transforming growth factor β1 through activation of endoplasmic reticulum stress and the unfolded protein response. Sci. Rep. 2016, 6, 22487.
- Choi, S.-H.; Hwang, S.B. Modulation of the Transforming Growth Factor-β Signal Transduction Pathway by Hepatitis C Virus Nonstructural 5A Protein. J. Biol. Chem. 2006, 281, 7468–7478.
- Naba, A.; Clauser, K.R.; Hoersch, S.; Liu, H.; Carr, S.A.; Hynes, R.O. The Matrisome: In Silico Definition and In Vivo Characterization by Proteomics of Normal and Tumor Extracellular Matrices. Mol. Cell. Proteom. 2012, 11.
- Naba, A.; Clauser, K.R.; Whittaker, C.A.; Carr, S.A.; Tanabe, K.K.; Hynes, R.O. Extracellular matrix signatures of human primary metastatic colon cancers and their metastases to liver. BMC Cancer 2014, 14, 518.
- Decaris, M.L.; Emson, C.L.; Li, K.; Gatmaitan, M.; Luo, F.; Cattin, J.; Nakamura, C.; Holmes, W.E.; Angel, T.E.; Peters, M.G.; et al. Turnover Rates of Hepatic Collagen and Circulating Collagen-Associated Proteins in Humans with Chronic Liver Disease. PLoS ONE 2015, 10, e0123311.
- Asselah, T.; Bièche, I.; Laurendeau, I.; Paradis, V.; Vidaud, D.; Degott, C.; Martinot, M.; Bedossa, P.; Valla, D.; Vidaud, M.; et al. Liver Gene Expression Signature of Mild Fibrosis in Patients with Chronic Hepatitis C. Gastroenterology 2005, 129, 2064–2075.
- Yasui, Y.; Abe, T.; Kurosaki, M.; Matsunaga, K.; Higuchi, M.; Tamaki, N.; Watakabe, K.; Okada, M.; Wang, W.; Shimizu, T.; et al. Non-invasive liver fibrosis assessment correlates with collagen and elastic fiber quantity in patients with hepatitis C virus infection. Hepatol. Res. 2019, 49, 33–41.
- Bracht, T.; Schweinsberg, V.; Trippler, M.; Kohl, M.; Ahrens, M.; Padden, J.; Naboulsi, W.; Barkovits, K.; Megger, D.A.; Eisenacher, M.; et al. Analysis of Disease-Associated Protein Expression Using Quantitative Proteomics—Fibulin-5 is Expressed in Association with Hepatic Fibrosis. J. Proteome Res. 2015, 14, 2278–2286.
- Nielsen, M.J.; Veidal, S.S.; Karsdal, M.A.; Ørsnes-Leeming, D.J.; Vainer, B.; Gardner, S.D.; Hamatake, R.; Goodman, Z.D.; Schuppan, D.; Patel, K. Plasma Pro-C3 (N-terminal type III collagen propeptide) predicts fibrosis progression in patients with chronic hepatitis C. Liver Int. 2015, 35, 429–437.
- Murawaki, Y.; Ikuta, Y.; Koda, M.; Kawasaki, H. Serum Type III Procollagen Peptide, Type IV Collagen 7S Domain, Central Triple-Helix of Type IV Collagen and Tissue Inhibitor of Metalloproteinases in Patients with Chronic Viral Liver Disease: Relationship to Liver Histology. Hepatology 1994, 20, 780–787.
- Valva, P.; Casciato, P.; Carrasco, J.M.D.; Gadano, A.; Galdame, O.; Galoppo, M.C.; Mullen, E.; De Matteo, E.; Preciado, M.V. The Role of Serum Biomarkers in Predicting Fibrosis Progression in Pediatric and Adult Hepatitis C Virus Chronic Infection. PLoS ONE 2011, 6, e23218.
- Lichtinghagen, R.; Michels, D.; Haberkorn, C.; Arndt, B.; Bahr, M.; Flemming, P.; Manns, M.P.; Boeker, K.H. Matrix metalloproteinase (MMP)-2, MMP-7, and tissue inhibitor of metalloproteinase-1 are closely related to the fibroproliferative process in the liver during chronic hepatitis C. J. Hepatol. 2001, 34, 239–247.
- Ljumovic, D.; Diamantis, I.; Alegakis, A.K.; Kouroumalis, E.A. Differential expression of matrix metalloproteinases in viral and non-viral chronic liver diseases. Clin. Chim. Acta 2004, 349, 203–211.
- Martinez-Castillo, M.; Hernandez-Barragan, A.; Flores-Vasconcelos, I.; Galicia-Moreno, M.; Rosique-Oramas, D.; Perez-Hernandez, J.L.; La Tijera, F.H.-D.; Montalvo-Jave, E.; Torre-Delgadillo, A.; Cordero-Perez, P.; et al. Production and activity of matrix metalloproteinases during liver fibrosis progression of chronic hepatitis C patients. World J. Hepatol. 2021, 13, 218–232.
- Murawaki, Y.; Ikuta, Y.; Kawasaki, H. Clinical usefulness of serum tissue inhibitor of metalloproteinases (TIMP)-2 assay in patients with chronic liver disease in comparison with serum TIMP. Clin. Chim. Acta 1999, 281, 109–120.
- Dudás, M.J.; Kovalszky, I.; Gallai, M.; Nagy, M.J.O.; Schaff, Z.; Knittel, T.; Mehde, M.; Neubauer, K.; Szalay, F.; Ramadori, G. Expression of Decorin, Transforming Growth Factor-beta1, Tissue Inhibitor Metalloproteinase 1 and 2, and Type IV Collagenases in Chronic Hepatitis. Am. J. Clin. Pathol. 2001, 115, 725–735.
- Ramnath, D.; Irvine, K.M.; Lukowski, S.W.; Horsfall, L.U.; Loh, Z.; Clouston, A.D.; Patel, P.J.; Fagan, K.J.; Iyer, A.; Lampe, G.; et al. Hepatic expression profiling identifies steatosis-independent and steatosis-driven advanced fibrosis genes. JCI Insight 2018, 3.
- Dong, C.; Li, H.-J.; Chang, S.; Liao, H.-J.; Zhang, Z.-P.; Huang, P.; Tang, H.-H. A Disintegrin and Metalloprotease with Thrombospondin Motif 2 May Contribute to Cirrhosis in Humans through the Transforming Growth Factor-β/SMAD Pathway. Gut Liver 2013, 7, 213–220.
- Kühn, J.; Gressner, O.A.; Götting, C.; Gressner, A.M.; Kleesiek, K. Increased serum xylosyltransferase activity in patients with liver fibrosis. Clin. Chim. Acta 2009, 409, 123–126.
- Gressner, O.A.; Gao, C. Monitoring fibrogenic progression in the liver. Clin. Chim. Acta 2014, 433, 111–122.
- Zhu, Z.-W.; Friess, H.; Wang, L.; Abou-Shady, M.; Zimmermann, A.; Lander, A.D.; Korc, M.; Kleeff, J.; Büchler, M.W. Enhanced glypican-3 expression differentiates the majority of hepatocellular carcinomas from benign hepatic disorders. Gut 2001, 48, 558–564.
- Llovet, J.M.; Chen, Y.; Wurmbach, E.; Roayaie, S.; Fiel, M.I.; Schwartz, M.; Thung, S.N.; Khitrov, G.; Zhang, W.; Villanueva, A.; et al. A Molecular Signature to Discriminate Dysplastic Nodules from Early Hepatocellular Carcinoma in HCV Cirrhosis. Gastroenterology 2006, 131, 1758–1767.
- Wurmbach, E.; Chen, Y.-B.; Khitrov, G.; Zhang, W.; Roayaie, S.; Schwartz, M.; Fiel, I.; Thung, S.; Mazzaferro, V.; Bruix, J.; et al. Genome-wide molecular profiles of HCV-induced dysplasia and hepatocellular carcinoma. Hepatology 2007, 45, 938–947.
- Clemente, M.; Nunez, O.; Lorente, R.; Rincon, D.; Matilla, A.; Salcedo, M.; Catalina, M.V.; Ripoll, C.; Iacono, O.L.; Banares, R.; et al. Increased intrahepatic and circulating levels of endoglin, a TGF-β1 co-receptor, in patients with chronic hepatitis C virus infection: Relationship to histological and serum markers of hepatic fibrosis. J. Viral Hepat. 2006, 13, 625–632.
- Guéchot, J.; Laudat, A.; Loria, A.; Serfaty, L.; Poupon, R.; Giboudeau, J. Diagnostic accuracy of hyaluronan and type III procollagen amino-terminal peptide serum assays as markers of liver fibrosis in chronic viral hepatitis C evaluated by ROC curve analysis. Clin. Chem. 1996, 42, 558–563.
- Karsdal, M.A.; Daniels, S.J.; Nielsen, S.H.; Bager, C.; Rasmussen, D.G.K.; Loomba, R.; Surabattula, R.; Villesen, I.F.; Luo, Y.; Shevell, D.; et al. Collagen biology and non-invasive biomarkers of liver fibrosis. Liver Int. 2020, 40, 736–750.
- Taleb, R.S.Z.; Moez, P.; Younan, D.; Eisenacher, M.; Tenbusch, M.; Sitek, B.; Bracht, T. Quantitative proteome analysis of plasma microparticles for the characterization of HCV-induced hepatic cirrhosis and hepatocellular carcinoma. Proteom. Clin. Appl. 2017, 11.
- El-Karef, A.; Kaito, M.; Tanaka, H.; Ikeda, K.; Nishioka, T.; Fujita, N.; Inada, H.; Adachi, Y.; Kawada, N.; Nakajima, Y.; et al. Expression of large tenascin-C splice variants by hepatic stellate cells/myofibroblasts in chronic hepatitis C. J. Hepatol. 2007, 46, 664–673.
- Benbow, J.H.; Elam, A.D.; Bossi, K.L.; Massengill, D.L.; Brandon-Warner, E.; Anderson, W.E.; Culberson, C.R.; Russo, M.W.; Delemos, A.S.; Schrum, L.W. Analysis of Plasma Tenascin-C in Post-HCV Cirrhosis: A Prospective Study. Dig. Dis. Sci. 2018, 63, 653–664.
- Choi, S.S.; Claridge, L.C.; Jhaveri, R.; Swiderska-Syn, M.; Clark, P.; Suzuki, A.; Pereira, T.A.; Mi, Z.; Kuo, P.C.; Guy, C.D.; et al. Osteopontin is up-regulated in chronic hepatitis C and is associated with cellular permissiveness for hepatitis C virus replication. Clin. Sci. 2014, 126, 845–855.
- Urtasun, R.; Lopategi, A.; George, J.; Leung, T.-M.; Lu, Y.; Wang, X.; Ge, X.; Fiel, M.I.; Nieto, N. Osteopontin, an oxidant stress sensitive cytokine, up-regulates collagen-I via integrin αVβ3 engagement and PI3K/pAkt/NFκB signaling. Hepatology 2011, 55, 594–608.
- Hackl, N.J.; Bersch, C.; Feick, P.; Antoni, C.; Franke, A.; Singer, M.V.; Nakchbandi, I.A. Circulating fibronectin isoforms predict the degree of fibrosis in chronic hepatitis C. Scand. J. Gastroenterol. 2009, 45, 349–356.
- Bracht, T.; Mölleken, C.; Ahrens, M.; Poschmann, G.; Schlosser, A.; Eisenacher, M.; Stühler, K.; Meyer, H.E.; Schmiegel, W.H.; Holmskov, U.; et al. Evaluation of the biomarker candidate MFAP4 for non-invasive assessment of hepatic fibrosis in hepatitis C patients. J. Transl. Med. 2016, 14, 201.
- Mölleken, C.; Ahrens, M.; Schlosser, A.; Dietz, J.; Eisenacher, M.; Meyer, H.E.; Schmiegel, W.; Holmskov, U.; Sarrazin, C.; Sorensen, G.L.; et al. Direct-acting antivirals-based therapy decreases hepatic fibrosis serum biomarker microfibrillar-associated protein 4 in hepatitis C patients. Clin. Mol. Hepatol. 2019, 25, 42–51.
- Castilla, A.; Prieto, J.; Fausto, N. Transforming Growth Factors β1 and α in Chronic Liver Disease. N. Engl. J. Med. 1991, 324, 933–940.
- Kinnman, U.A.N. In Situ Expression of Transforming Growth Factor-ß1?3, Latent Transforming Growth Factor-ß Binding Protein and Tumor Necrosis Factor-a in Liver Tissue from Patients with Chronic Hepatitis C. Scand. J. Gastroenterol. 2000, 35, 1294–1300.
- Divella, R.; Daniele, A.; Gadaleta, C.; Tufaro, A.; Venneri, M.T.; Paradiso, A.; Quaranta, M. Circulating Transforming Growth Factor-β and Epidermal Growth Factor Receptor as Related to Virus Infection in Liver Carcinogenesis. Anticancer Res. 2012, 32, 141–145.
- Xu, L.; Hui, A.Y.; Albanis, E.; Arthur, M.J.; O’Byrne, S.M.; Blaner, W.S.; Mukherjee, P.; Friedman, S.L.; Eng, F.J. Human hepatic stellate cell lines, LX-1 and LX-2: New tools for analysis of hepatic fibrosis. Gut 2005, 54, 142–151.
- Florimond, A.; Chouteau, P.; Bruscella, P.; Le Seyec, J.; Mérour, E.; Ahnou, N.; Mallat, A.; Lotersztajn, S.; Pawlotsky, J.-M. Human hepatic stellate cells are not permissive for hepatitis C virus entry and replication. Gut 2015, 64, 957–965.
- Aoudjehane, L.; Bisch, G.; Scatton, O.; Granier, C.; Gaston, J.; Housset, C.; Roingeard, P.; Cosset, F.-L.; Perdigão, F.; Balladur, P.; et al. Infection of Human Liver Myofibroblasts by Hepatitis C Virus: A Direct Mechanism of Liver Fibrosis in Hepatitis C. PLoS ONE 2015, 10, e0134141.
- Devhare, P.B.; Sasaki, R.; Shrivastava, S.; Di Bisceglie, A.M.; Ray, R.; Ray, R.B. Exosome-Mediated Intercellular Communication between Hepatitis C Virus-Infected Hepatocytes and Hepatic Stellate Cells. J. Virol. 2017, 91.
- Bartosch, B. Piecing together the key players of fibrosis in chronic hepatitis C: What roles do non-hepatic liver resident cell types play? Gut 2014, 64, 862–863.
- Foschi, F.G.; Domenicali, M.; Giacomoni, P.; Dall’Aglio, A.C.; Conti, F.; Borghi, A.; Bevilacqua, V.; Napoli, L.; Mirici, F.; Cucchetti, A.; et al. Is there an association between commonly employed biomarkers of liver fibrosis and liver stiffness in the general population? Ann. Hepatol. 2020, 19, 380–387.
- Meissner, E.G.; McLaughlin, M.; Matthews, L.; Gharib, A.M.; Wood, B.J.; Levy, E.; Sinkus, R.; Virtaneva, K.; Sturdevant, D.; Martens, C.; et al. Simtuzumab treatment of advanced liver fibrosis in HIV and HCV-infected adults: Results of a 6-month open-label safety trial. Liver Int. 2016, 36, 1783–1792.
- Roehlen, N.; Crouchet, E.; Baumert, T.F. Liver Fibrosis: Mechanistic Concepts and Therapeutic Perspectives. Cells 2020, 9, 875.


