Chronic infection by the hepatitis C virus (HCV) is a major cause of liver diseases, predisposing to fibrosis and hepatocellular carcinoma. Liver fibrosis is characterized by an overly abundant accumulation of components of the hepatic extracellular matrix, such as collagen and elastin, with consequences on the properties of this microenvironment and cancer initiation and growth. This review will provide an update on mechanistic concepts of HCV-related liver fibrosis/cirrhosis and early stages of carcinogenesis, with a dissection of the molecular details of the cross-talk during disease progression between hepatocytes, the extracellular matrix and hepatic stellate cells.
- liver fibrosis
- cirrhosis
- chronic hepatitis C
- carcinogenesis
- extracellular matrix
1. Introduction
|
Virus |
HBV |
HCV |
|---|---|---|
|
Viral family |
Hepadnaviridae |
Flaviviridae |
|
Genome |
DNA and cccDNA |
RNA |
|
Life cycle |
Genome integration, expression of HBx protein, insertional activation of cellular oncogenes, cccDNA (minichromosome) |
Exclusively cytoplasmic |
|
Persistence |
Nucleus-located cccDNA |
Chronic inflammation, oxidative stress, alterations in cellular signaling and metabolism |
2. Main Actors of Liver Fibrosis
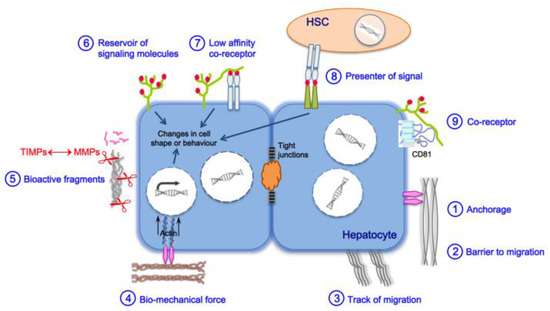
3. Liver Fibrosis and Cirrhosis
3.1. General Pan-Etiology Features
3.2. Features Linked to HCV Pathogenesis
|
HCV Proteins |
ECM Proteins or Cytokines |
|---|---|
|
Capsid core |
LOX ∞ [61] Procollagen I ∞ [62] Collagen I ∞ [61] MMP-2 ∞ [58] |
|
MMP-9 ∞ [63] |
|
|
COX-2 ∞ [63] |
|
|
Syndecan-1 * [31] |
|
|
Thrombospondin-1 ∞ [61] |
|
|
CTGF ∞ [58] |
|
|
TGF-β2 ◊ [67] |
|
|
Endoglin ∞ [68] |
|
|
Envelope glycoproteins E1 and/or E2 |
Glypican-3 * [69] |
|
TGF-β1 ◊ [66] |
|
|
Cysteine autoprotease NS2 |
MICA ∞ [70] TGF-β2 ◊ [67] |
|
Serine protease and helicase NS3 |
Procollagen I ∞ [62] MMP-9 ∞ [71] |
|
COX-2 ∞ [71] |
|
|
Thrombospondin-1 [72] |
|
|
Osteopontin * [64] |
|
|
TGF-β type I receptor * [73] |
|
|
NS3 with its cofactor NS4A |
MMP-9 ∞ [71] |
|
COX-2 ∞ [71] MICA ∞ [74] |
|
|
NS4B |
MMP-2 ∞ [76] |
|
NS5A |
MMP-2 ∞ [63] |
|
MMP-9 ∞ [63] |
|
|
COX-2 ∞ [63] |
|
|
Thrombospondin-1 ∞ [72] |
|
|
Osteopontin * [64] |
|
|
RNA-dependent RNA polymerase NS5B |
Osteopontin * [64] |
|
MICA ∞ [70] TGF-β ◊ [75] |
|
ECM Proteins/Cytokine |
F0/F1 |
F2 |
F3 |
F4 |
HCC |
References |
|---|---|---|---|---|---|---|
|
Collagens I, III, V |
F1 |
|||||
|
Collagen XII |
||||||
|
Collagen XIV |
||||||
|
Collagen XVI |
[59] |
|||||
|
Collagen XVIII |
[59] |
|||||
|
PIIINP |
F1 |
|||||
|
MMP-2, -7, -9 |
F1 |
|||||
|
TIMP-1 |
||||||
|
ADAM-TS1 |
[93] |
|||||
|
ADAM-TS2 |
[94] |
|||||
|
Xylosyltransferase-2 |
F1 |
|||||
|
Glypican-3 |
||||||
|
Hyaluronic acid |
||||||
|
Decorin |
F1 |
[92] |
||||
|
Biglycan |
[59] |
|||||
|
Fibromodulin |
[60] |
|||||
|
Lumican |
||||||
|
Versican |
F1 |
|||||
|
Tenascin-C |
||||||
|
Osteopontin |
F1 |
|||||
|
Fibronectin |
||||||
|
Fibronectin isoforms |
[108] |
|||||
|
Elastin |
||||||
|
MFAP-4 † |
F1 |
|||||
|
Fibulin-5 |
[84] |
|||||
|
TGF-β1 (protein, mRNA) |
||||||
|
TGF-β1 (serum levels) |
F1 |
|||||
|
TGF-β2 |
F1 |
F0 |
[67] |
|||
|
Endoglin (protein, serum levels) |
[100] |
|||||
|
Endoglin (mRNA) § |
[68] |
a Color codes: green, upregulation; dark green: higher upregulation; blue, downregulation; dark blue: higher downregulation; grey, no change; magenta, no correlation with liver fibrosis stage. † MFAP-4, microfibrillar-associated protein-4 (associated with elastin fibers). § Endoglin mRNA was found upregulated in chronically HCV-infected patients compared to noninfected patients but not correlating with liver fibrosis stage.
3.2.1. Collagens and derived fragments.
By specifically analyzing the ECM components of biopsies from HCV-infected patients with liver fibrosis, levels of collagens type I, III and V were found increased from stages F0-F1 to F4 [59][79,80]. However, this is a hallmark of all fibrotic diseases [102]. These fibrillar collagens form arrays incorporating fibril-associated collagens with interrupted triple helices (FACIT), such as collagens XII and XIV [114]. Hepatic collagen XII amounts decreased with increasing stages of HCV-related fibrosis, whereas collagen XIV levels increased [59,84]. Collagen XVI, another FACIT not associated to collagens I, III, or V fibrils [115], was found underexpressed, specially at the cirrhotic stage of HCV-related fibrosis [59]. Collagen XVII is a transmembrane protein and a main component of hemidesmosomes, which are cell-ECM junctions anchoring epithelial cells to the basement membrane by interacting with integrins outside and with intermediate filaments of the cytoskeleton inside the cells [116]. Patients with advanced fibrosis/cirrhosis linked to chronic hepatitis C revealed focal positivity for collagen XVII in sinusoidal lining cells and few cholangiocytes, a pattern not observed in patients with cholangitis-linked cirrhosis [47]. Collagen XVII was also observed in the cytoplasm of HPC, and in the surrounding basal membrane in end-stage HCV-linked fibrosis. Collagen XVIII is a basement membrane collagen almost exclusively expressed in the liver, and hepatocytes were identified as its main source [28,117]. It can be cleaved at its C-terminus to generate endostatin, a powerful anti-angiogenic agent [117][118]. Collagen XVIII was found underexpressed in HCV-diseased liver [59], and through the subsequent down-expression of endostatin, hepatocytes could play an unforeseen role in neo-angiogenesis during hepatic neoplasia.
Studies attempting to define etiology-dependent molecular signatures of liver fibrosis identified serum collagen-derived biomarkers that variably evolve during chronic hepatitis B and C. Pro-C3 (N-terminal type III collagen propeptide), a fragment of collagen III formation, has been proposed as a serum predictive biomarker of fibrosis progression in patients with chronic hepatitis C [85]. These patients displayed higher serum levels of this biomarker than HBV-chronically infected patients [50]. By contrast, serum of HBV-infected patients had higher levels of the protease-cleaved 7S domain of the amino-terminal propeptide of type IV procollagen P4NP7S, a biomarker of type IV collagen formation. Chronic HBV-infected patients also had higher serum levels of markers of collagens III, IV and VI degraded by matrix metalloproteases than chronic HCV patients, as a reflection of a greater basement remodeling induced by chronic hepatitis B [50].
3.2.2. Enzymes of the ECM (Table 3).
3.2.2.1. Lysyl oxidases.
The lysyl oxidase family of enzymes comprises 5 members: LOX and LOX-like 1 to 4 (LOXL). They are copper-dependent secreted amine oxidases that cross-link monomers of collagen or elastin, to form insoluble fibrils. Hepatocytes of healthy livers do not express, or express only low amounts of lysyl oxidases [119]. In viral hepatitis C, LOX and LOXL1 expression is strongly enhanced in activated HSCs but not in hepatocytes [119]. Likewise, in viral hepatitis, collagen deposits are only observed in fibrotic zones, not around hepatocytes, in contrast to what is observed in fibrotic liver diseases of other etiologies. LOXL2 is strongly induced in fibrotic liver, where it localizes to regions of collagenous matrix and to a-SMA-positive fibroblasts-like cells [120]. More specifically, in fibrotic liver diseases related to active hepatitis C infection, LOX and LOXL2 were detected at the fibrotic disease interface composed of fibroblasts, hepatocytes and neo-vasculature [120]. LOX was found to contribute to collagen stabilization in liver fibrosis, promote fibrogenic activation of HSCs, and limit fibrosis reversal [121]. LOXL2 also mediates fibrotic matrix stabilization, and stimulates differentiation of HPC toward fibrogenic cholangiocytes [122]. The fibrosis-promoting activities of LOX and LOXL2 are susceptible to occur even after cessation of the chronic liver injury. Together with the global morphological reorganization of the liver due to ECM accumulation that alters liver metabolism, these impaired enzymatic activities might contribute to the persistence of clinical signs of liver disease after SVR in the case of chronic hepatitis C, in the absence of any therapy targeting the enzymes [120][123]. This feature may be even more pronounced when alcohol intake and/or metabolic syndrome complicate chronic infection. Thrombospondin-1 belongs to the family of matricellular proteins, which mediate cell-cell and cell-ECM interactions but are not primary structural ECM elements. It plays a role in collagen homeostasis, through its binding to fibrillar collagens, pro-LOX, matrix metalloproteases (MMPs) and TGF-b1 [71,72]. The core protein of HCV was found to downregulate LOX, while up-regulating the genes of collagen I (COL1A1) and thrombospondin-1 (THBS1) [73]. Similar gene expression profiles were obtained from mice xenograft tumors derived from HCV-infected human hepatocytes [74]. Mechanistically, HCV, and core in particular, activate the production of latent TGF-b1 by infected hepatocytes [53][73,74]. After its secretion in the microenvironment, latent TGF-b1 is then cleaved into active TGF-b1 by thrombospondin-1, the expression of which is upregulated by HCV [75], and the core protein in particular [73]. Active TGF-b1 finally activates hepatic stellate cells, which contribute to fibrogenesis by (over) producing ECM components [73]. Interestingly, THBS1 is a TGF-b1 target gene, which creates a vicious loop where activated HSCs are committed to ECM overproduction.
3.2.2.2. Matrix Metalloproteases (MMPs) and their inhibitors.
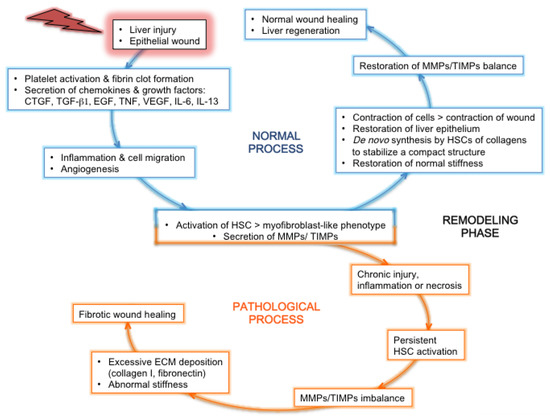
Under physiological conditions of liver regeneration during wound healing, the normal equilibrium between MMPs and their inhibitors (tissue inhibitors of MMPs, TIMPs) is restored, whereas the MMP/TIMP ratio remains imbalanced during fibrosis (Figure 2). The mRNA and protein expression of several MMPs is modified during chronic hepatitis C: the gelatinases MMP-2 and -9 are enhanced [57,58,76], while the collagenases MMP-1 and -13 are down-regulated [57]. A recent study reported increased serum levels of MMP-2, -7 and -9 in patients with chronic hepatitis C, which correlated to fibrosis stage for MMP-7 [90]. Gelatinases contribute to the degradation of the basement membrane collagen IV, while collagenases such as MMP-1 and -13 break down interstitial collagens I and III. During chronic hepatitis C, the upregulation of gelatinases will lead to a major reshuffling of the basement membrane, with the destruction of the structural support of cells, and the down-regulation of collagenases will amplify the deposition of insoluble collagen fibers and aggravate the fibrotic phenotype. As mentioned earlier, hepatic collagen XIV is increased during HCV-related fibrosis [59,84]; since this collagen is collagenase-sensitive, this observation is in line with the down-regulation of collagenases [57].
In parallel, expression and secretion of TIMPs, in particular TIMP-1, are enhanced to compensate for an increased activity of MMPs [57,124]. Elevated levels of MMP-2 and -9 were reported in the serum and/or liver specimens of chronic hepatitis C patients, correlating with the fibrosis stage but not with the viral load [63,76][88]. The serum level of TIMPs was suggested as an indicator of hepatic fibrosis: increased serum levels of TIMP-1 in chronic hepatitis C patients correlated with the stage of liver fibrosis [86], better than those of TIMP-2 [91]. Interestingly, serum levels of MMP-9 and TIMP-1 decreased after SVR in fibrotic HCV patients treated with DAA, in parallel with the regression of fibrosis [125]. When fibrosis etiologies are compared, it appears that MMP-10 and -11 are upregulated at late fibrosis stages of chronic hepatitis B, whereas at similar stages of chronic hepatitis C, MMPs-2 and -9 are upregulated [89]. In NAFLD and non-alcoholic steatohepatitis, MMP-9 and -10 were found at significantly higher levels than in chronic viral hepatitis B and C [89].
To study the underlying mechanisms of such enzyme involvement in HCV-related fibrogenesis, hepatoma cell lines stably expressing the HCV protein(s) of interest were generated. The highest levels of MMP-2 were secreted when primary HSCs were grown with hepatoma cells expressing HCV core, compared with HSCs grown alone or in the presence of “regular” hepatoma cells [58]. Hepatocytes expressing HCV core secrete high amounts of TGF-b1, and exhibit a transcriptional upregulation of the connective tissue growth factor (CTGF), a mitogen stimulating the production of ECM components and enzymes by HSCs. This suggests that HCV core is a key regulator of MMP(-2) expression, possibly through a TGF-b1-mediated upregulation of CTGF [58]. Along similar lines, Chang liver cells expressing HCV core displayed elevated levels of MMP-9 transcript and protein, not observed in NS5A-expressing hepatocytes [63]. This suggests a direct effect of the core protein on the regulation of MMP-9 protein synthesis, and could explain the elevated levels of hepatic MMP-9 in biopsies from patients with HCV, not observed in patients with non-alcoholic steatohepatitis. Interestingly, both core- and NS5A-expressing cells displayed elevated levels of cyclooxygenase-2 (COX-2), a key enzyme of the biosynthesis of prostaglandins, mediators of inflammation. High hepatic levels of COX-2 were also reported in HCV-patients biopsies, which places core and NS5A at a regulatory hub between inflammation and fibrogenesis [63]. This is reminiscent of a strong correlation between the inflammatory activity of the liver of fibrotic HCV patients and the transcriptional levels of TIMP-1 [88]. Similar higher levels of MMP-9 were reported in the serum of chronically HCV-infected patients, and in HCV-infected hepatoma cells than in uninfected patients or cells, and MMP-9 displayed a greater enzymatic activity [71]. COX-2 was also found overexpressed in HCV-infected hepatocytes. Mechanistic studies revealed a regulatory role of the HCV serine protease NS3, together with its cofactor NS4A, underlying such over-expression and -activity: HCV NS3 activated the ERK1/2 – p38 pathway, leading to the phosphorylation of ERK1/2 and p38. This led to nuclear factor-kB (NF-kB) activation, and translocation to the nucleus where it promoted MMP9 transcription, leading to MMP-9 (over)expression [83]. In parallel, NF-kB promoted COX2 transcription, and once biosynthesized, COX-2 translocated to the nucleus where it acted as a transcriptional promoter of MMP9. COX-2 and MMP-9 contributed to inflammation and fibrosis through the synthesis of prostaglandins and ECM degradation, respectively [71]. As noticed earlier, HCV-mediated fibrosis appears tightly linked to liver inflammation, with on one hand viral proteins (core, NS3, NS5A) acting as modulators of inflammation pathways, and on the other hand inflammation mediators such as COX-2 acting as regulators of liver fibrosis. This is also in line with the anatomical observations of gathering of mononuclear cells at the hepatic lobules in HCV-related fibrosis, a hallmark of inflammatory activity not observed in alcohol-related fibrosis [46], and the presence of prominent aggregates of lymphocytes in peri-portal zones [46][47]. The non structural protein NS4B was also identified as an activator of MMP-2 expression, both at the mRNA and protein levels [76]. Mechanistic studies revealed that NS4B activated the ERK/JNK pathway, which engaged the transcription factor STAT3, thus contributing to MMP2 transcription.
3.2.2.3. A disintegrin and metalloprotease with thrombospondin motifs (ADAM and ADAM-TS).
ADAMs, comprising ADAM-TS, are membrane-bound or secreted zinc-proteases of the ECM, with a broad tissue distribution. Through their disintegrin and metalloprotease domains, they are therefore able to carry out cell adhesion and protease activities, respectively. In the liver, they are involved in the regulation of epithelial cell regeneration after liver injury, and able to directly degrade ECM components, thereby promoting ECM rearrangement during wound healing and fibrosis. A strong correlation between ADAM-9, -28, -TS1 vs MMP-2 and a-SMA was identified in biopsies from patients with diseased liver of any etiologies [126]. Also no differences in ADAM expression were detected in biopsies of different etiologies. The expression of ADAM-10 and -17 correlated with the severity of the fibrosis in patients with chronic liver diseases [127]. Higher expression of ADAM-TS2 was detected in cirrhotic than in normal liver, and correlated with TGF-b1 expression [94], independently of the etiology of the liver disease. However, ADAM-TS2 expression is indirectly linked to HCV-induced fibrogenesis. In an attempt to identify non invasive markers of liver fibrosis during chronic hepatitis C, the ELF (Enhanced Liver Fibrosis) test was put forward; it evaluates the serum levels of TIMP1, HA, N-terminal peptide of procollagen type III (PIIINP), and includes age for statistical robustness [128]. This test has been shown to properly identify moderate and severe fibrosis in the context of chronic hepatitis C [128], and in particular the serum levels of PIIINP could differentiate between mild and moderate/high HCV-related liver fibrosis [85]. Since ADAM-TS2 excises the N-propeptide of the fibrillar procollagens types I, II, III and V, one can infer that an implicit evaluation of ADAM-TS2 expression and activity is contained in the ELF test, in link with HCV pathogenesis and fibrogenesis.
Recent genome-wide association studies, aimed at identifying genetic polymorphism behind the risk of developing HCV-related HCC, also unveiled a mechanistic scheme linking ADAMs to HCV pathogenesis. A single nucleotide polymorphism (SNP) was identified in the 5’-flanking region of MICA (Major Histocompatibility Complex class I-related chain A, or MHC class I polypeptide-related sequence A) as a susceptibility gene for HCV-induced HCC. In these patients, this SNP was associated with progression from chronic hepatitis C to cirrhosis and HCC [129]. MICA is a cell surface glycoprotein of epithelial and endothelial cells, monocytes and fibroblasts, involved in particular in antiviral defense responses [130]. It functions as an indicator of cellular stress and activates circulating cytotoxic natural killer (NK) cells, key actors of immune surveillance, deploying in particular anticancer activity. MICA expression is induced in cells undergoing oncogenic or viral stress [131]. In the context of HCV infection, membrane-bound MICA (mMICA) expression in HCV-infected hepatocytes was downregulated by the NS3/4A serine protease [74]. Hepatocytes expressing the cysteine protease NS2 or the polymerase NS5B exhibited decreased MICA expression when co-cultured with NK cells [70], supporting the notion that HCV NS2 and NS5B disable MICA in infected hepatocytes, thereby inhibiting the ability of these cells to respond to stimuli from NK cells. MICA is shed from the membrane by ADAM-9, -10 and -17, which generates soluble MICA (sMICA) [132][133,134]. ADAM-mediated shedding of MICA therefore leads to higher levels of sMICA and lower levels of mMICA. Since ADAM-9, -10 and -17 expression increases with the severity of fibrosis in patients with chronic liver diseases [127][131], more MICA shedding occurs in these pathologies, translated into higher levels of sMICA. Interestingly, sMICA is an inhibitor of the activity of NK cells, in particular their activity against HCC [135]. Elevated levels of sMICA were observed in patients with advanced HCC, associated with impaired activation of NK cells [131]. In the peculiar context of HCV-induced HCC, patients bearing the allele at higher risk of HCC occurrence displayed higher sMICA levels [129]. This correlated with a poorer prognosis, and a higher risk of developing the liver disease in a context of escape from NK-mediated immune surveillance [129,131]. Finally, a correlation was found between high sMICA levels, the higher risk allele and the development of HCC in HCV-infected cirrhotic patients who failed to develop a SVR [136]. Of note, therapies combining DAA and inhibitors of ADAMs are currently debated for this type of patients [131,135].
3.2.3. Proteoglycans (Table 3).
Proteoglycans (PGs) are membrane-associated and soluble proteins of the ECM. They are composed of long chains of sugar molecules, anchored on a short polypeptide chain: sugars can represent up to 90% of their weight. This is in contrast to matrix glycoproteins, comprising small sugar chains anchored to a long polypeptide chain, and where sugars do not represent more than 60% of their weight (see below ¶ 3.2.5.). Through their GAG chains, PGs interact with numerous regulatory molecules, such as cytokines, growth factors, hormones. This has repercussions on a myriad of normal and pathological cellular processes. PGs are classified as SLIPs (Syndecan-like integral membrane PGs), GRIPs (Glypican-related integral membrane heparan sulfate PGs), membrane-associated b-glycan and CD44, extracellular SLRPs (Small leucine-rich PGs) and hyalectans [137].
3.2.3.1. Membrane-associated PGs.
The SLIP family of PGs comprises four molecules, syndecans-1 to 4. Syndecan-1 harbors heparan and chondroitin sulfate GAGs (HS and CS, respectively), while other syndecans only bear HS. Interestingly, syndecan-1 and -4 were identified as co-factors of HCV attachment and entry into hepatocytes [31,138,139]. Mechanistically, syndecan-1 forms with the tetraspanin CD81 a complex internalized with virions during viral entry. HCV core and syndecan-1 colocalized during the intracellular trafficking of virions [31]. When chronic infection of hepatoctytes was established, syndecan-1 expression was down-regulated [31]. This is in line with observations on biopsies collected from patients with HCC, including HCV-infected patients [140]: the down-regulation was more pronounced in the tumoral tissue than in peripheral non-tumoral zones, and correlated with the aggressiveness of the tumor. However, these data remain controversial, since other studies reported an up-regulation of syndecan-1 during cirrhosis, and in association with HCV-related HCC [141]. Syndecan-1 down-regulation correlated with the up-regulation of xylosyltransferase-2 [31], a key enzyme located in the ER/Golgi compartments, involved in the biosynthesis of the tetrasaccharide linkage region of CS and HS PGs [142]. Xylosyltransferase-2 activity was found increased in the serum of patients with HCV-related liver fibrosis at early stages [95,96], evocative of a dramatic remodeling of PGs within the fibrotic ECM. Of note, this correlation became negative in the cirrhotic range of liver fibrosis [95]. Thus, higher enzymatic activity at the fibrotic stage could be seen as a compensatory mechanism against HCV-mediated liver injury; at the cirrhotic stage, as more damages have occurred, this compensation might become inefficient or disappear.
The GRIP family of PGs comprises the glypicans, anchored to the membrane through a glycosylphosphatidylinositol tail [143]. Among the 6 glypicans, the most studied is glypican-3 (GPC3), an oncofetal protein widely expressed during development, but silenced in adult tissues. It is seen as a negative regulator of cell growth [144], and it is overexpressed in cancers such as embryonic tumors [145], and HCC [97]. In a cohort of HCV-infected patients with liver disease, high levels of GPC3 transcript and protein appeared as good markers of early stages of HCC, discriminating from dysplastic nodules [98]. Moreover, GPC3 was also strongly upregulated in early and advanced HCC, compared with normal tissue. As described for syndecan-1, GPC3 forms a complex with CD81. Mechanistic studies revealed the following loops: in resting liver, GPC3 is associated to CD81 at the plasma membrane, thereby preventing CD81 to bind the cytosolic transcriptional repressor hematopoietically-expressed Homeobox (Hhex); this repressor therefore migrates to the nucleus, where it exerts its growth inhibitory role. In regenerating liver (notably after an injury), GPC3 binding to CD81 decreases, which leads to an increased binding of CD81 with Hhex in the cytosol; less Hhex therefore migrates to the nucleus, and proliferation is boosted [146]. During HCV-mediated liver disease and HCC, the virus, and in particular its membrane glycoprotein E2 which binds CD81, was shown to mimic GPC3 in infected hepatocytes, thereby interfering with the GPC3/CD81 binding [69]. The decrease of GPC3/CD81 entities leads to more CD81/Hhex cytosolic binding, so less nuclear Hhex. This fuels neoplastic proliferation. HCV is thus likely to enhance liver neoplasia by acting as a growth promoter of neoplastic hepatocytes, through its binding to CD81 to the detriment of GPC3 [69].
A remarkable feature of SLIPs and GRIPs is the ability of their ectodomain to be cleaved by ECM proteases, in a process called shedding. The resulting soluble domain then acts as an autocrine and/or paracrine signal in the microenvironment, per se and via the factors attached to its GAG chains. Elevated serum levels of shed syndecan-1 have been proposed as biomarkers of prediction of liver fibrosis, but not of liver inflammation in chronic hepatitis C patients [147]. Although of note, these data should be cautiously taken, since high serum levels of syndecan-1 are also observed during e.g. inflammation, sepsis, and vascular dysfunctions. Concerning GPC3, plasma levels of the shed form were found higher in patients with HCC than in healthy individuals; they were also higher in patients with HCC induced by HCV than by HBV or other etiologies [148]. The full length form of GPC3 also assayed in blood samples correlated with tumoral GPC3 expression only in patients with HCV-induced liver carcinoma. GPC3 has now demonstrated its added value as a biomarker of HCC, and has become a target of cancer immunotherapy [149]. Indeed T cells expressing GPC3-specific chimeric antigen receptor (CAR-T) were generated, to target and destroy GPC3-positive HCC. Several clinical trials are currently ongoing to evaluate the safety and tolerability of such an immune strategy (e.g. clinicaltrials.gov identifiers NCT02395250, NCT02905188). Elevated serum levels of these shed PGs are an indication of increased shedding, and therefore of increased systemic exposure to the factors retained in the GAG moieties, such as TGF-bs. These cytokines could then exert their fibrogenic and oncogenic effect at a distance from their site of biosynthesis. GPC3 was found to suppress the expression and signalling of TGF-b2, which led to activation of cell growth and cell cycle progression [150]; strategies aimed to suppress GPC3 expression were thus proposed as valuable options for the clinical management of GPC3-positive HCC patients. Concerning syndecan-1, paradoxical data were reported: indeed, retention of TGF-b1 and thrombospondin-1 in its GAG chains was shown to inhibit the conversion of latent TGF-b1 into its active form, thereby decreasing the availability of active TGF-b1 [151]. Due to increased shedding, shed syndecan-1 at higher amounts would enter the circulation together with its latent TGF-b1 cargo. Less activation and availability of TGF-b1 would then translate into less activation of HSCs, and therefore less synthesis of fibrogenic molecules (see also above ¶ 3.2.2.1.). This is somewhat conflictual with the correlation found between high serum levels of shed syndecan-1 and the prediction of liver fibrosis in chronic hepatitis C patients [147]. However, the precise mechanistic link between HCV pathogenesis and increased PG shedding is still missing, although the HCV-induced enhancement of protease expression could play a role (see above ¶ 3.2.2.).
The cluster of differentiation molecule 44 (CD44) is a membrane-associated adhesion glycoprotein of the ECM, considered as a “part-time PG” since covalent attachment of a CS or HS to its protein chain depends on developmental or pathological cues [152]. It is produced as standard (CD44s) and variant (CD44v) isoforms, and it is the receptor of hyaluronan (HA), a soluble non-sulfated GAG (see above ¶ 2.). Specific interactions between CD44 and HA are of major importance for the maintenance of the proper (stem cell) niche properties under physiological conditions, and of the biology of cancer stem cells under pathological circumstances [152]. Together with other cell surface molecules, CD44 was proposed as a marker of cancer stem cells in HCC, which are responsible for tumor progression and aggressiveness. In particular, CD44s expression in HCC was related to the TGF-b-mediated regulation of the epithelial-to-mesenchymal transition [153], and its level of expression in tumoral tissue was associated with negative prognosis [154]. In HCV-induced HCC, the variant 9 of CD44 was identified as a biomarker of liver cancer stem cells, and its expression correlated with tumor invasiveness and poor patient survival [155]. Interestingly, CD44v9 expression also correlated with the expression of markers of liver fibrosis (fibronectin) and of HSC activation (myosin light chain), suggesting that CD44v9 could play a role at early stages of the HCV-induced liver disease. In patients with chronic hepatitis C, high plasma levels of HA were associated to the progression of liver fibrosis, whereas low serum levels of the interferon gamma-inducible protein 10 (IP-10) correlated with a favorable outcome to anti-HCV therapy [156]. IP-10 is a biomarker of liver inflammatory activity, correlating with lobular fibrosis and necroinflammation, but not with portal inflammation or fibrosis. From these observations, Matsuura and coworkers identified a mechanistic loop involving CD44. HCV-infected hepatocytes were found to secrete high amounts of IP-10, in response to the engagement and activation of the innate immune Toll-like receptor 2 (TLR2). This secretion depended upon the stimulation of TLR2 by HA, and induced an enhancement of CD44 expression. HA was then also able to bind CD44, and to further stimulate the production of IP-10. Additionally, CD44 was found to directly interact with TLR2 through its extracellular domain. The mechanistic loop is therefore as follows: high amounts of endogenous HA generated during HCV infection stimulate TLR2 in HCV-infected hepatocytes, which induces the production of the pro-inflammatory factor IP-10; IP-10 in turn stimulates the expression of CD44 which, as a receptor of HA and a co-receptor of TLR2, will enhance the cellular response to HA stimulation in a vicious loop aggravating the inflammatory profile [156]. Moreover, CD44 acted in synergy with TGF-b1 at early stages of cell transformation, and to confer stem properties to transformed cells [153].
Another mechanistic loop underlying HCV pathogenesis and involving CD44 was recently described [64]. HCV replication in hepatocytes induced the endogenous expression of osteopontin, a matricellular protein. Osteopontin then localized at the ER, where it contributed to HCV replication, assembly and infectivity by direct binding to the HCV proteins core, NS3, NS5A, and NS5B. A pool of osteopontin was also processed by proteolysis inside HCV-infected hepatocytes, and secreted in the microenvironment where it exerted autocrine and paracrine signalling, through integrin aVb3 and CD44 binding. This latter binding activated the focal adhesion kinase which contributed to HCV replication, and to HCV assembly. Further data indicate that HCV replication was particularly enhanced in liver cancer stem cells expressing CD44 and EpCAM (epithelial cell adhesion molecule), and that osteopontin also activated HCV replication in these cells [65]. It did so by regulating the stemness of the CD44+/EpCAM+ cells, thereby inactivating interferon signalling and fuelling HCV replication. CD44 is therefore a major actor of HCV pathogenesis, and together with its ligand osteopontin, emerges as a key regulator of cancer stem cells and HCV replication [65]. Recent studies dealing with CD44 also revealed that HCV would replicate at higher rates in liver cancer stem cells than in differentiated hepatocytes, which is of uttermost importance for the comprehension of HCV-induced liver disease, notably HCC.
3.2.3.2. Soluble extracellular PGs.
These PGs are classified in extracellular SLRPs, and pericellular PGs and hyalectans. SLRPs are extracellular PGs, and represent the largest family of PGs, characterized by a small core protein comprised of a central region of leucine-rich repeats [143]. Most SLRP members carry chondroitin-, dermatan- or keratan-sulfate chains, but a few lack any GAG. Main SLRPs comprise biglycan, decorin, asporin, fibromodulin, and lumican. Pericellular PGs such as the heparan sulfate PGs perlecan and agrin are main components of basement membranes; in the liver, they surround blood vessels and the biliary compartment (mature bile ducts and the HPC niche) in portal zones [143],[157,158]. They bind collagens (specially type IV), fibronectin and laminins (see below ¶ 3.2.5.), thereby assisting collagen fibrillogenesis, and playing a key role in ECM stabilization and in the maintenance of the functional status of mature connective tissues. Hyalectans (a.k.a. lecticans) are components of the pericellular or interstitial matrix, residing in close vicinity of the cell membrane, important for the communication between cells and ECM as well as for the regulation of proliferation and migration. They form a ternary binding complex with HA and the matricellular protein tenascin-R, which creates large aggregates in the ECM. The four members of the hyalectan family are versican, aggrecan, brevican and neurocan [143][159].
The SLRP biglycan is involved in collagen fibril assembly, in an ECM-bound form. After proteolytic cleavage, it is released as a soluble form. Proteomic analyses of the ECM of liver biopsies of chronic hepatitis C patients with liver fibrosis revealed a downregulation of the ECM-bound form of biglycan [59]. However, serum levels were found not significantly altered in HCV patients compared to normal controls [160]. Biglycan is a CD44 ligand, and also binds the innate immune receptors TLR2 and TLR4 [161]. It is generally considered as a pro-inflammatory molecule, however its role and the explanation behind its downregulation during HCV-induced liver disease remain unclear. As biglycan, decorin plays a role in collagen fibrillogenesis (its name comes from the fact that it “decorates” collagen I fibrils). It binds growth factors, notably TGF-b1. The Glisson’s capsule of the liver is strongly enriched in decorin. During HCV-induced liver disease, decorin deposition was identified as an early and sensitive indicator of active ECM remodeling after liver injury [92]. This feature correlated with increased TGF-b1 expression, for low to moderate stages of fibrosis. However, in this study, a comparable pattern was observed for liver disease induced by other etiologies. As biglycan, decorin is able to modulate TLR2 and TLR4-mediated signalling, and play a role in inflammation; however, our knowledge about decorin and (HCV-induced) liver fibrosis remains piecemeal [162]. Fibromodulin and lumican are SLRPs decorated with keratan sulfate GAG chains, and participate in collagen fibril assembly. Both molecules are biosynthesized by HSCs, and are weakly expressed in normal liver [60]. Fibromodulin transcript and protein were found overexpressed in patients with HCV-induced cirrhosis, in parallel with collagen I expression; however, in this study, a similar overexpression was observed for other etiologies of liver fibrosis. Mechanistic analyses revealed that the overproduction of fibromodulin was under the control of oxidative stress, locally induced by the liver injury [60] (and by HCV-induced liver injury in particular [163]). Lumican was also found overexpressed in liver fibrosis [102], and in HCV-induced fibrosis in particular, on liver biopsies [84] and specifically in the ECM [59]. Lumican expression correlated with the severity of the (HCV-related) fibrosis [102][84][81], and high levels of lumican were reported in the plasma of HCV-positive patients with liver disease [84][81]. However, the exact roles of fibromodulin and lumican in (HCV-related) liver disease remain to be determined.
Of the four hyalectans, only versican exhibits a wide distribution; brevican and neurocan are only expressed in neural tissue, and aggrecan in cartilage. Versican biosynthesis is stimulated by TGF-β1 and PDGF [143]. In the ECM, it interacts with matricellular proteins, fibronectin and chemokines via its core domain or GAG chains. It also binds CD44, integrin β1, and at the surface of macrophages, toll-like receptors; versican is therefore involved in key cellular functions such as adhesion, proliferation, migration and invasion. It has been reported as a good biomarker of advanced liver fibrosis in patients with HCV infection or NAFLD, since its hepatic mRNA and serum levels were higher at advanced stages of the disease than at earlier ones [93]. Concomitantly, the levels of ADAM-TS1, involved in versican proteolysis, were enhanced, and higher in liver fibrosis with than without steatosis [134]. Extracellular vesicles are nano‐sized particles shed into body fluids by many cell types that carry various bioactive molecules; among them, plasma microvesicles (100-1000 nm in size) are key messengers of cellular communication. Enhanced microvesicle levels were linked to disease activity and progression, e.g. microvesicles isolated from patients with HCV-related decompensated cirrhosis induce vascular hypocontractility, contributing to portal hypertension and circulatory dysfunctions [164]. Versican-positive microvesicles were found elevated in patients with HCV-induced cirrhosis, and further increased with HCC [103]. In patients who had received DAA, the levels of versican-positive microvesicles dropped at the end of treatment and remained low throughout the 48-week follow‐up [164]. This supports the view that DAA‐induced eradication of HCV could promote a reversal of fibrosis, and versican-positive microvesicles could be a potential early biomarker of liver fibrosis.
3.2.4. Matricellular proteins (Table 3).
These proteins are non-structural components of the ECM, in contrast to structural elements such as collagens and fibronectin. As their name indicate, they form a bridge between the matrix (matri-) and the cells (-cellular). They bind to other ECM proteins and cell surface receptors, growth factors, cytokines and MMPs, thereby modulating the activity and accessibility of these factors, and mediating enzymatic activities to regulate ECM homeostasis [165]. Main members of this family are thrombospondins, tenascins, osteopontin (a.k.a. SPP1) and CCNs [named from the first 3 proteins identified in this family: Cysteine-rich angiogenic inducer 61 (CYR61), CTGF, and Nephroblastoma overexpressed protein (NOV)]. Notably, CCN2 is also known as the connective tissue growth factor (CTGF) [166]. Increased serum levels of CYR61 or CCN1 were proposed as biomarkers of HCV-induced HCC post-SVR [14] (see ¶ 1). The link between HCV pathogenesis and thrombospondin-1 has already been described in ¶ 3.2.2.1.
3.2.4.1 Tenascins.
There are four tenascins, C, R, W and X, but only tenascins-C and -X are ubiquitously expressed. Tenascin-C is present in all organs during fetal development, but weakly expressed in normal adult tissue. During mechanical stress or pathological tissue remodeling, such as wound healing, fibrogenesis or tumorigenesis, its expression is enhanced. A chronic liver injury leads to the activation of HSCs, with concomitant enhanced secretion of tenascin-C in chronic hepatitis C [104]. Serum tenascin-C has therefore been suggested as a biomarker to discriminate fibrotic/cirrhotic patients with active hepatitis C from healthy controls, and from those with HCV eradication after antiviral therapy [105]. Tenascin-C serum levels were found good indicators of ongoing hepatic injury and inflammation in fibrotic/cirrhotic patients. After SVR, serum levels of this protein returned to the baseline observed in healthy individuals, suggesting the reversion of HSCs to their quiescent state [105]. More specifically, serum levels of large splice variants of tenascin-C were proposed as useful markers of the inflammatory activity during chronic hepatitis C, and in particular of the degree of piecemeal necrosis [167]. However, somewhat discordant results were reported, since tenascin was found under-expressed in the liver ECM of chronically HCV-infected patients, in correlation with the fibrosis stage [59]. In fine, no mechanistic hypothesis was put forward concerning the link between HCV infection, liver disease and tenascin-C expression.
3.2.4.2. Osteopontin.
Osteopontin or SPP1 (secreted phosphoprotein 1) is an adhesive phosphorylated acidic glycoprotein, existing as a full length form and cleaved fragments. It is produced by HSCs, cholangiocytes, hepatocytes, HPC [168], macrophages and T helper lymphocytes, and its production is enhanced by PDGF and inflammation. Its serum levels are viewed as markers of liver fibrosis from several etiologies (including chronic hepatitis C), significantly correlating with the fibrosis stage, liver insufficiency, portal hypertension, and the presence of HCC [160,169,170][171]. In patients with chronic hepatitis C, osteopontin gene expression enhancement was part of a gene expression signature of moderate (F2) compared to mild (F1) fibrosis [82]. Mechanistic studies revealed a vicious loop linking osteopontin to HCV replication and pathogenesis: endogenous or exogenous osteopontin favors viral replication in hepatocytes [106], as well as assembly and infectivity of viral particles [64]. It likely does so through direct interactions with viral structural and non-structural proteins (core, NS3, NS5A, NS5B), in the ER and at the membrane of lipid droplets, both sites of HCV replication and assembly. Osteopontin is cleaved intracellularly, and released fragments are secreted in the extracellular space, where they bind CD44 and the aVb3 integrin in an autocrine and paracrine manner [64]. These cues stimulate HCV replication in the ER through the activation of the focal adhesion kinase (FAK), and promote HCV assembly at lipid droplets. In turn, HCV indirectly stimulates the endogenous production of osteopontin, notably through the activation of oxidative stress [172] and of cellular kinases (MAPK, JNK, PI3-K, MEK1/2). Therefore a vicious circle is generated. Further investigations revealed that osteopontin enhanced HCV replication in a peculiar population of hepatic cells, i.e. CD44+ cancer stem cells [65]. Interestingly, osteopontin was described as a regulator of stemness of these cells, which are among the targets of HCV infection in the liver, and the cells of origin of liver cancers (HCC or intra-hepatic cholangiocarcinoma) [173]. This can be linked to the observation that increased serum levels of osteopontin unfavorably correlated with the early recurrence of HCV-related HCC [174]. Osteopontin was also described as an inducer of ductular reactions [172]. In the recent study of ductular reactions in chronic liver disease of various etiologies [47], HPC (stem cells) from ductular reactions of patients with chronic and active hepatitis C displayed gene signatures related to metabolism and hepatocytes, with gene networks enriched for cell movement and receptor activity, not observed in patients with cholangitis. Osteopontin is therefore at a pivotal position: as a regulator of stemness of cells targeted by HCV during infection - in link with its activity as an inducer of ductular reactions - and as an enhancer of infection. Along similar lines, HPC play a key role in HCV pathogenesis, since they are HCV target cells and cells of origin of liver cancers. A more specific link between osteopontin, stemness, ductular reactions and fibrosis occurrence was established [172]: osteopontin-/- transgenic mice less readily developed chemically-induced liver fibrosis than wild type animals, suggesting that osteopontin expression is increased after liver injury. Osteopontin of the diseased liver ECM then decreased hepatocyte proliferation, induced stem cell expansion and ductular reactions, and concomitantly, fueled the up-regulation of collagen I in HSCs via its binding to aVb3 and CD44, contributing to an enhanced fibrogenic response [107,172] together with the modulation of TGF-b signaling [168]. Another vicious circle is therefore unraveled: osteopontin is directly involved in the initial response to the hepatic insult, and promotes ductular reactions, which contribute to sustain injury and to liver fibrosis.
Lastly, a genetic link between osteopontin and chronic hepatitis C was discovered; indeed, studies of genetic polymorphism revealed SNPs in the promoter region of osteopontin in chronic hepatitis C patients. In particular, the SNP at nucleotide -443 (C or T) showed an association with the activity of hepatitis C. This activity was defined as the serum levels of alanine aminotransferase (ALT): low (ALT > 30 IU/l), medium (30 < ALT < 80 IU/l) or high (ALT > 80 IU/l). The frequency of T/T homozygotes prevailed in the medium- and high-activity groups, whereas C/T heterozygotes prevailed in the low-activity group [175].
3.2.4.3. CCN2 or CTGF.
CCN2 or CTGF is a mitogen and a matricellular protein expressed by hepatocytes, HSCs, vascular endothelial cells and cholangiocytes [96][176], and overexpressed in chronic liver diseases [177]. CTGF binds to sulfated GAGs and HSPGs (Figure 1), thereby acting as a local reservoir that can attract competent cells to fibrotic sites. It also plays an autocrine role on HSCs, leading to abundant production of ECM molecules. Elevated serum and liver levels of this cytokine were found in fibrotic/cirrhotic patients chronically infected with HCV [177,178]. However CTGF levels did not correlate with the stage of fibrosis, and were higher in patients with progressive fibrosis than in those with end-stage cirrhotic liver disease [176]. HCV induced CTGF overexpression, which in turn stimulated the production of procollagen-I by HSCs, in a TGF-β1-dependent manner [57,58][179]. The viral protein core was suggested as a main trigger of CTGF overproduction [58].
3.2.5. Adhesive glycoproteins.
The main components of this family of ECM proteins are fibronectin, laminins and nidogens. They are formed of small sugar chains anchored to long polypeptide chains, and organised as structural modules enabling a great diversity of protein-protein and ECM-cell interactions. This modular organisation contributes to ECM cohesion. Fibronectin is the main component of the space of Disse in normal liver; it is produced at high rates during liver fibrosis. Fibronectin is found in the plasma as its full length form produced by hepatocytes, and as isoforms produced by activated HSCs, defined by the presence of alternatively spliced domains [fibronectin containing extra-domains A or B (EDA or EDB), variable domain]. This variable domain is also found glycosylated at a specific threonine residue, known as the oncofetal fibronectin (oFN) isoform [108]. Laminins and nidogens are main components of basement membranes, involved in the formation of a dense network of ECM proteins. Under physiological conditions, laminins are absent from the space of Disse, but present in the basement membrane that surrounds bile ducts and are in contact with HPC [180].
3.2.5.1. Fibronectin.
Circulating levels of the fibronectin isoforms were analyzed in patients with liver fibrosis related to chronic hepatitis C or to other etiologies [108]. Total fibronectin did not correlate with any parameter in either group. However, levels of fibronectin isoforms taken individually were significantly higher in patients with chronic hepatitis C compared to healthy controls, and high levels of EDA and oFN correlated with high scores of liver fibrosis (F2 to F4). Additionally, low levels of both isoforms were associated with the absence of significant liver fibrosis. By contrast, in patients with liver disease unrelated to HCV, none of the isoforms correlated with any parameter. When examining specific aspects of the fibrosis, it was found that EDA elevation significantly correlated with necrosis, and oFN predicted inflammation [181]. A slightly better correlation was found when combining oFN plasma levels with those of some elements of the complement system. Similarly the combination of oFN with a fraction of the complement correlated better with the fibrosis score than oFN alone [181].
3.2.5.2. Laminin and nidogen.
As described above, HPC produce during fibrosis an excess of fibrogenic mediators (TGF-β1 and -β2, PDGF, CTGF), which fuel HSC proliferation and activation. In an attempt to understand how the niche of HPC influences their behaviour, Lorenzini et al. analyzed the liver tissue from various liver injury models and compared it with healthy tissue, and addressed the functional significance of the laminin - HPC interaction. In liver tissue from patients with chronic hepatitis C, a close contact between HPC, myofibroblasts and laminin was observed [182]. Laminin, produced by myofibroblasts and HPC, deposited as a sheath surrounding HPC, which contributed to the cohesion of the niche around HPC. Laminin also deposited in the space of Disse, in a phenomenon called capillarization of sinusoids observed in liver disease related to chronic hepatitis C, but not to other etiologies [183]. In vitro experiments revealed that HPC grown on laminin-enriched ECM maintained their stemness properties, while HPC grown on fibronectin differentiated into hepatocytes. Laminin strongly repressed the expression of C/EBPa, an early hepatocyte gene encoding a liver-selective transcription factor [182]. Laminin therefore contributed to maintain HPC in their undifferentiated status, which could fuel the deregulation of HPC proliferation underlying liver cancer.
Nidogen was found expressed on cell membranes of biliary epithelia, and not on hepatocytes in normal liver tissue. In HCV-infected diseased tissue, nidogen staining was seen on cell membranes of periportal hepatocytes [183]. However no functional analysis was performed, which could explain such location.
3.2.6. Elastin.
Elastin is a minor ECM component in normal liver, which nevertheless plays a key role to confer strength and elasticity to the organ. It is cross-linked from tropoelastin by LOX to give rise to insoluble elastin fibers, in a similar reaction as collagen fibrillogenesis [24,27]. Elastin fibers are stable components of the ECM, although proteolytically processed by MMP-2, -9 and the elastase MMP-12. TGF-β1-mediated activation of HSCs leads to increased expression of tropoelastin [184], and the deposition of insoluble elastic fibers in the diseased liver could lead to irreversibility of fibrosis [27]. Elastin was found overexpressed in fibrotic and cirrhotic livers, compared to normal organ. This overexpression correlated with the severity of the hepatic disease [102]. In liver biopsies from patients with chronic hepatitis C, increasing amounts of elastin correlated with the severity of liver fibrosis [84][83]. This was accompanied by an increase of expression of fibulin-5 and microfibrillar-associated protein-4, proteins associated to elastic fibers and involved in elastogenesis [59][185,186]. More selectively, microfibrillar-associated protein-4 was found overexpressed in liver biopsies [84], more specifically in the ECM fraction of liver biopsies [59], and also at high levels in the serum of HCV-positive patients [84], correlating with the severity of the disease [109,110]. Mechanistically, elastin processed into small peptide fragments acts as a chemo-attractant for cells of the immune system that will locally secrete inflammatory cytokines (Figure 1). Elastin peptides can also stimulate the proliferation of activated HSCs and their secretion of MMP-12 [27], thereby aggravating the imbalance between MMP and TIMP, and contributing to fibrogenesis (Figure 2).
3.2.7. TGF-b and HCV pathogenesis (Table 3).
In most organs, such as the liver, TGF-βs are secreted as a large latent TGF-β complex, formed by the mature TGF-β protein non-covalently attached to the precursor protein (latency-associated peptide), linked to large latent TGF-β-binding proteins (LTBPs) [187]. LTBPs play a key role in the activation of this latent complex, by targeting it to the cell surface followed by its secretion into extracellular areas, where activation likely occurs, notably via thrombospondin-1 (see above [61], ¶ 3.2.2.) and furin [72][188]. Of the three TGF-β isoforms (TGF-β1, -β2 and -β3) described in mammals, TGF-β1 is the most extensively studied in human liver diseases. Under normal conditions, TGF-β1 is mainly secreted from Kupffer cells, while hepatocytes only secrete small amounts [72]. Upon any liver injury in general, and chronic hepatitis C in particular, high levels of TGF-β1 and -β2 are secreted in the space of Disse by hepatocytes and neighboring cells [57,66], fueling fibrogenesis [34,189] (Figure 2), whereas TGF‐β3 was reported as an anti-fibrotic cytokine in the liver. Indeed, TGF-β3 was shown to inhibit TGF-β1 expression at the transcriptional level, and suppress collagen synthesis [190]. TGF-β1 and -β2 secretion contributes to HSC activation and transformation into myofibroblast-like cells [66]. Indeed TGF-β1-mediated activation of HSCs [191] results in: (i) vitamin A loss accompanied by a decrease in serum retinoid levels [192]; (ii) α-SMA expression [42,58,61]; (iii) greater TGF-β1 secretion [72], in a vicious loop. TGF-βs are therefore key regulators of fibrosis, involved in chronic liver diseases, which contribute to all disease stages, from initial liver injury to fibrosis, cirrhosis, and HCC [193].
In patients with chronic hepatitis C, increased serum levels of TGF-β1 and -β2 were reported [87][67], together with high amounts of TGF-β1 and -β2 mRNA in liver specimens [194], compared to healthy individuals. These correlated with the degree of liver fibrogenesis but not with HCV RNA levels [67][111,195]. Higher circulating levels were also found in patients with HCC, compared to patients with liver cirrhosis, and these levels were the highest when the liver disease was caused by HCV infection or HBV/HCV co-infection, compared to other etiologies [113]. TGF-β1 mRNA in liver biopsies strongly correlated with procollagen type I mRNA, as a further indication of the link between TGF-β1 expression and liver fibrosis/cirrhosis [111]. LTBP-1 and TGF-β1 protein levels were found upregulated in liver tissue from patients with chronic hepatitis C [112,195]. Analyses selectively performed on the ECM fraction of liver biopsies revealed large deposits of LTBP-1 and -4 in HCV patients at stage F3, suggestive of a pivotal role of the large latent TGF-β complex at the F2-F3 transition [59]. The mechanistic crosstalk between HCV and TGF-β synthesis and activity has been investigated in various in vitro or in vivo models of chronic HCV infection. HCV-infected hepatocytes implanted into mice generated nodular tumors enriched in cancer stem-like cells. These hepatocytes were shown to recruit activated murine fibroblasts into the xenograft stroma, by secretion of TGF-β1 [194]. This led to stromal changes, such as activation of HSCs (a-SMA expression and enhanced synthesis of MMP-2), and appearance of markers of tumor-associated fibroblasts: collagen I, CTGF, vimentin, fibroblast-specific protein 1. HCV infection also resulted in overproduction of TGF-β1 in liver cells isolated from patients with chronic hepatitis C [66]. HCV core, expressed in human hepatoma cells, strongly enhanced TGF-β1 mRNA expression in these cells, and upregulated TGF-β1 promoter [196]. The MAPK pathway was found involved in the engagement of TGF-β1 by the HCV core protein. Other studies have similarly addressed the crosstalk between this viral protein and TGF-β1-mediated fibrogenesis, as described earlier in link with enzymes of the ECM (¶ 3.2.2.1. and 3.2.2.2.). The expression of HCV core, E1 and E2 proteins in human hepatocytes revealed that core and E2 were responsible for the enhanced secretion of TGF-β1, through the overproduction of the glucose-regulated protein 94 (GRP94), a chaperone protein of the ER lumen and an ER stress marker [66]. This further emphasizes the link between TGF-β1 production, and oxidative and ER stress, all induced by HCV [72][77]. GRP94 upregulation would engage the NF-κB pathway, which would in turn trigger the enhancement of TGF-β1 production. The exogenous addition of the HCV NS3 protease to cultured hepatocytes led to increased expression of collagen I, TGF-β1 and TGF-β type I receptor [73] ; additionally, NS3 directly interacted with the TGF-β type I receptor at the surface of HCV-infected cells, and in HCV-infected chimeric mice, an anti-NS3 antibody attenuated HCV-induced liver fibrosis. HCV NS3 present in extracellular areas, possibly arising from the leakage from injured hepatocytes, would therefore function via its direct binding to the TGF-β type I receptor and its activation, thereby enhancing liver fibrosis [73]. Moreover, the functionally-active protease domain of NS3 was found required for TGF-β1 activity, i.e. for the activation of the TGF-β1 promoter [72]. This implies the presence of the cofactor NS4A with NS3. NS3-4A could then enhance the proteolytic activity of thrombospondin-1 and furin for the cleavage of the latent TGF-β complex and subsequent activation of TGF-β. NS3-4A also interacted with SMURF2, a negative regulator of the TGF-β signaling. Through this interaction, NS3-4A blocked the negative regulation of TGF-β signaling, thereby enhancing the cellular response to TGF-β [75]. Accordingly, hepatocytes expressing either non-structural proteins (NS3 to NS5B) or NS3-4A of HCV, once stimulated by TGF-β, exhibited a pro-tumoral transcriptional program, with enhanced expression of genes involved in cell proliferation, negative regulation of cell differentiation, epithelial-to-mesenchymal transition and vasculature development [75], all genes related to carcinogenesis. Expressing HCV NS5A into hepatocytes also induced the production of high levels of TGF-β1, and NS5A was found important for the activation of the TGF-β1 promoter [72]. Interestingly, NS5A associates with the membrane of the ER [197], where it contributes to ER stress and induces an ER-to-nucleus signal transduction pathway involving NF-kB [77][198]. This pathway could contribute to chronic inflammation and liver fibrosis. Again, this confirms the link between HCV infection, virus-induced ER stress, and TGF-β1 overexpression and activity. As already noted for NS3, NS5A directly interacted with the TGF-β type I receptor at the surface of cells expressing NS5A [78], and together with other viral proteins, might therefore contribute to liver fibrogenesis through the engagement of TGF-β signaling.
TGF‐β2 was found upregulated by the co-expression of two proteins of HCV, core and NS2, most markedly in hepatocytes compared to HSCs and Kupffer cells [67]. Mechanistically, HCV-induced ER stress following hepatocyte infection caused the translocation of the liver-enriched and hepatocyte-specific transcriptional regulator cAMP responsive element binding protein H (CREBH) from the ER to the Golgi apparatus, and its subsequent activation. The activated CREBH was then transported to the nucleus, where it targeted and bound the DNA CREBH response element and the CRE responsive element-like sequence. Both elements modulate the expression of their target gene, TGFβ2 [190]. Through this pathway, HCV therefore enhanced TGF-β2 expression in hepatocytes [67]. After its release in the extracellular milieu, TGF-β2 exerted effects on hepatocytes and HSCs in autocrine and paracrine manners, increasing the expression of profibrotic molecules (TGF-β1 and -β2, α-SMA, collagen I), thereby promoting liver fibrosis [67].
Endoglin is a transmembrane glycoprotein belonging to the TGF-β receptors family, where it constitutes, together with betaglycan, the TGF-β type Ⅲ receptor family. Endoglin interacts with the TGF-β receptors types I and II, and acts as a key switch by producing different variant forms, adjusting ligand affinity, and creating links with a versatile receptor network, thereby modulating the specific outcome of TGF-β-dependent and -independent pathways [199]. It is expressed as a dimer at the surface of proliferating vascular endothelial cells, of quiescent and activated HSCs, but not of hepatocytes. It binds TGF-β1 and -β2, and in HSCs, it is associated to the TGF-β type Ⅱ receptor, and shows highest expression during maximal cell activation. It is activated by phosphorylation through the activity of the TGF-β type Ⅱ receptor part. Its expression is upregulated during liver damage and transiently induced in HSC by TGF-β1. Circulating levels of endoglin were higher in patients with chronic hepatitis C than in healthy individuals [100], and correlated with the severity of the liver disease, with highest values in HCV-positive cirrhotic patients. Intra-hepatic levels of endoglin were also higher in HCV-infected livers than in uninfected biopsies [68,100], as also observed for TGF-β1 levels. Endoglin expression in the liver correlated with the METAVIR score. This suggests that increased levels of released TGF-β1, linked to HCV-induced liver fibrosis/cirrhosis, may be responsible for the endoglin upregulation, in a positive feedback loop where the ligand (TGF-β1) stimulates the expression of (one of) its receptor (endoglin). This could possibly create a vicious circle, with a sustained or continuous activation of the TGF-β receptors, leading to the production of overly abundant components of the ECM by activated HSCs, and therefore to fibrosis. This poses endoglin as a key element of HCV pathogenesis, playing a crucial role in liver fibrogenesis. Mechanistic studies revealed that endoglin upregulation in HCV-infected hepatocytes was mainly due to the HCV core protein [68]. In these cells, core-induced overexpression of endoglin contributed to the activation of the TGF-β signaling pathway, with the transcription of target genes involved in cell proliferation and acquisition of (cancer) stem cell properties. In an attempt to discover novel determinants of HCV-related liver fibrosis progression, a joint French/Swiss study group identified an endoglin variant, Thr5Met, the frequency of which was higher among HCV/fibrosis patients than among HCV/controls [200]. This variant was even depleted in patients without fibrosis. An additional SNP was found in a regulatory region of the endoglin gene, in link with increased risk of liver fibrosis in HCV patients, and predicted to decrease the DNA-binding affinity of HNF4a (involved in the expression of liver-specific genes) [200]. This would lead to a lower transcription of liver-specific genes, and, as mentioned earlier, to the maintenance of hepatic progenitors in their undifferentiated state, leading to the occurrence of (HCV-infected) cancer stem cells underlying HCC [47].
4. Are HSCs Direct Targets of HCV Infection?
5. Fibrosis Reversal in the Era of DAAs in HCV-Induced Liver Fibrosis
- Conclusion - Perspectives.
In the era of DAAs which raise hope of eradicating HCV, HCV infection still remains a leading cause of hepatic failure due to advanced liver disease and HCC, because curing the infection does not fully restore liver homeostasis. Furthermore, DAA treatment alone may not be sufficient for a complete cure of fibrosis, as several factors other than the virus contribute to liver deterioration. Lastly, patients under antiviral therapy variably respond to the regression of fibrosis. The mechanism of HCV-induced liver disease is a multifaceted process, since various host genes are altered, and since host cells respond to infection/viral components by mobilizing or producing enzymes, growth factors and chemokines which activate quiescent HSCs. HCV chronic infection leads to a deep remodeling of the entire liver ECM architecture, through direct interactions between viral, ECM and cellular proteins, and through indirect effects (e.g. promotion of oxidative and ER stress, of inflammation, of stemness). HCV-induced overexpression of TGF-b, the most potent profibrogenic cytokine, contributes to HCV replication, and to the activation of HSCs, the promotion of their survival, and the inhibition of HSC apoptosis, mechanisms by which liver disease progresses. Consequently, several general mechanisms involved in liver fibrosis/cirrhosis development contribute to tumorigenesis. TGF-b signaling facilitates HCV replication in hepatocytes, and could promote survival of pre-cancerous cells; furthermore, HCV replicates at higher rates in liver cancer stem cells.
Thus, efforts toward a deeper comprehension of host/virus/ECM interactions, and of the underlying mechanisms by which hepatic dysfunctions emerge, spread and persist after HCV infection are therefore still needed, in order to develop therapies that cure liver disease in addition to curing infection.References.
- Plummer, M.; de Martel, C.; Vignat, J.; Ferlay, J.; Bray, F.; Franceschi, S. Global Burden of Cancers Attributable to Infections in 2012: A Synthetic Analysis. Lancet Glob Health 2016, 4, e609-616, doi:10.1016/S2214-109X(16)30143-7.
- Villanueva, A. Hepatocellular Carcinoma. N Engl J Med 2019, 380, 1450–1462, doi:10.1056/NEJMra1713263.
- Mitchell, J.K.; Lemon, S.M.; McGivern, D.R. How Do Persistent Infections with Hepatitis C Virus Cause Liver Cancer? Curr Opin Virol 2015, 14, 101–108, doi:10.1016/j.coviro.2015.09.003.
- Lemon, S.M.; McGivern, D.R. Is Hepatitis C Virus Carcinogenic? Gastroenterology 2012, 142, 1274–1278, doi:10.1053/j.gastro.2012.01.045.
- WHO Combating Hepatitis B and C to Reach Elimination by 2030. Advocacy Brief 2016.
- Polaris Observatory HCV Collaborators Global Prevalence and Genotype Distribution of Hepatitis C Virus Infection in 2015: A Modelling Study. Lancet Gastroenterol Hepatol 2017, 2, 161–176, doi:10.1016/S2468-1253(16)30181-9.
- WHO | Hepatitis C Available online: http://www.who.int.gate2.inist.fr/mediacentre/factsheets/fs164/en/ (accessed on 29 January 2018).
- Hajarizadeh, B.; Grebely, J.; Dore, G.J. Epidemiology and Natural History of HCV Infection. Nat Rev Gastroenterol Hepatol 2013, 10, 553–562, doi:10.1038/nrgastro.2013.107.
- Nault, J.-C.; Colombo, M. Hepatocellular Carcinoma and Direct Acting Antiviral Treatments: Controversy after the Revolution. J. Hepatol. 2016, 65, 663–665, doi:10.1016/j.jhep.2016.07.004.
- Baumert, T.F.; Jühling, F.; Ono, A.; Hoshida, Y. Hepatitis C-Related Hepatocellular Carcinoma in the Era of New Generation Antivirals. BMC Med 2017, 15, 52, doi:10.1186/s12916-017-0815-7.
- Hamdane, N.; Jühling, F.; Crouchet, E.; El Saghire, H.; Thumann, C.; Oudot, M.A.; Bandiera, S.; Saviano, A.; Ponsolles, C.; Roca Suarez, A.A.; et al. HCV-Induced Epigenetic Changes Associated With Liver Cancer Risk Persist After Sustained Virologic Response. Gastroenterology 2019, 156, 2313-2329.e7, doi:10.1053/j.gastro.2019.02.038.
- Hengst, J.; Falk, C.S.; Schlaphoff, V.; Deterding, K.; Manns, M.P.; Cornberg, M.; Wedemeyer, H. Direct-Acting Antiviral-Induced Hepatitis C Virus Clearance Does Not Completely Restore the Altered Cytokine and Chemokine Milieu in Patients With Chronic Hepatitis C. J. Infect. Dis. 2016, 214, 1965–1974, doi:10.1093/infdis/jiw457.
- Akuta, N.; Suzuki, F.; Hirakawa, M.; Kawamura, Y.; Sezaki, H.; Suzuki, Y.; Hosaka, T.; Kobayashi, M.; Kobayashi, M.; Saitoh, S.; et al. Amino Acid Substitutions in Hepatitis C Virus Core Region Predict Hepatocarcinogenesis Following Eradication of HCV RNA by Antiviral Therapy. J. Med. Virol. 2011, 83, 1016–1022, doi:10.1002/jmv.22094.
- Takeda, H.; Takai, A.; Iguchi, E.; Mishima, M.; Arasawa, S.; Kumagai, K.; Eso, Y.; Shimizu, T.; Takahashi, K.; Ueda, Y.; et al. Oncogenic Transcriptomic Profile Is Sustained in the Liver after the Eradication of the Hepatitis C Virus. Carcinogenesis 2021, doi:10.1093/carcin/bgab014.
- Paul, D.; Madan, V.; Bartenschlager, R. Hepatitis C Virus RNA Replication and Assembly: Living on the Fat of the Land. Cell Host Microbe 2014, 16, 569–579, doi:10.1016/j.chom.2014.10.008.
- El Sebae, G.K.; Malatos, J.M.; Cone, M.-K.E.; Rhee, S.; Angelo, J.R.; Mager, J.; Tremblay, K.D. Single-Cell Murine Genetic Fate Mapping Reveals Bipotential Hepatoblasts and Novel Multi-Organ Endoderm Progenitors. Development 2018, 145, doi:10.1242/dev.168658.
- Lee, Y.A.; Wallace, M.C.; Friedman, S.L. Pathobiology of Liver Fibrosis: A Translational Success Story. Gut 2015, 64, 830–841, doi:10.1136/gutjnl-2014-306842.
- Rauterberg, J.; Voss, B.; Pott, G.; Gerlach, U. Connective Tissue Components of the Normal and Fibrotic Liver. I. Structure, Local Distribution and Metabolism of Connective Tissue Components in the Normal Liver and Changes in Chronic Liver Diseases. Klin. Wochenschr. 1981, 59, 767–779.
- Friedman, S.L. Hepatic Stellate Cells: Protean, Multifunctional, and Enigmatic Cells of the Liver. Physiol. Rev. 2008, 88, 125–172, doi:10.1152/physrev.00013.2007.
- De Minicis, S.; Seki, E.; Uchinami, H.; Kluwe, J.; Zhang, Y.; Brenner, D.A.; Schwabe, R.F. Gene Expression Profiles during Hepatic Stellate Cell Activation in Culture and in Vivo. Gastroenterology 2007, 132, 1937–1946, doi:10.1053/j.gastro.2007.02.033.
- Lin, X.Z.; Horng, M.H.; Sun, Y.N.; Shiesh, S.C.; Chow, N.H.; Guo, X.Z. Computer Morphometry for Quantitative Measurement of Liver Fibrosis: Comparison with Knodell’s Score, Colorimetry and Conventional Description Reports. J. Gastroenterol. Hepatol. 1998, 13, 75–80.
- Duarte, S.; Baber, J.; Fujii, T.; Coito, A.J. Matrix Metalloproteinases in Liver Injury, Repair and Fibrosis. Matrix Biology: Journal of the International Society for Matrix Biology 2015, 44–46, 147–156, doi:10.1016/j.matbio.2015.01.004.
- Rojkind, M.; Giambrone, M.A.; Biempica, L. Collagen Types in Normal and Cirrhotic Liver. Gastroenterology 1979, 76, 710–719.
- Kagan, H.M. Lysyl Oxidase: Mechanism, Regulation and Relationship to Liver Fibrosis. Pathol. Res. Pract. 1994, 190, 910–919, doi:10.1016/S0344-0338(11)80995-7.
- Martinez-Hernandez, A.; Amenta, P.S. The Extracellular Matrix in Hepatic Regeneration. FASEB J. 1995, 9, 1401–1410.
- Iozzo, R.V.; Schaefer, L. Proteoglycan Form and Function: A Comprehensive Nomenclature of Proteoglycans. Matrix Biol. 2015, 42, 11–55, doi:10.1016/j.matbio.2015.02.003.
- Kanta, J. Elastin in the Liver. Front Physiol 2016, 7, 491, doi:10.3389/fphys.2016.00491.
- Musso, O.; Rehn, M.; Saarela, J.; Théret, N.; Liétard, J.; Hintikka, null; Lotrian, D.; Campion, J.P.; Pihlajaniemi, T.; Clément, B. Collagen XVIII Is Localized in Sinusoids and Basement Membrane Zones and Expressed by Hepatocytes and Activated Stellate Cells in Fibrotic Human Liver. Hepatology 1998, 28, 98–107, doi:10.1002/hep.510280115.
- Ricard-Blum, S.; Vallet, S.D. Fragments Generated upon Extracellular Matrix Remodeling: Biological Regulators and Potential Drugs. Matrix Biol. 2019, 75–76, 170–189, doi:10.1016/j.matbio.2017.11.005.
- Sun, S.; Song, Z.; Cotler, S.J.; Cho, M. Biomechanics and Functionality of Hepatocytes in Liver Cirrhosis. J Biomech 2014, 47, 2205–2210, doi:10.1016/j.jbiomech.2013.10.050.
- Grigorov, B.; Reungoat, E.; Gentil Dit Maurin, A.; Varbanov, M.; Blaising, J.; Michelet, M.; Manuel, R.; Parent, R.; Bartosch, B.; Zoulim, F.; et al. Hepatitis C Virus Infection Propagates through Interactions between Syndecan-1 and CD81, and Impacts the Hepatocyte Glycocalyx. Cell. Microbiol. 2017, 19, doi:10.1111/cmi.12711.
- Wells, J.M.; Gaggar, A.; Blalock, J.E. MMP Generated Matrikines. Matrix Biol. 2015, 44–46, 122–129, doi:10.1016/j.matbio.2015.01.016.
- Schuppan, D.; Ashfaq-Khan, M.; Yang, A.T.; Kim, Y.O. Liver Fibrosis: Direct Antifibrotic Agents and Targeted Therapies. Matrix Biol. 2018, 68–69, 435–451, doi:10.1016/j.matbio.2018.04.006.
- Ricard-Blum, S.; Baffet, G.; Théret, N. Molecular and Tissue Alterations of Collagens in Fibrosis. Matrix Biol. 2018, doi:10.1016/j.matbio.2018.02.004.
- Jung, Y.; Witek, R.P.; Syn, W.-K.; Choi, S.S.; Omenetti, A.; Premont, R.; Guy, C.D.; Diehl, A.M. Signals from Dying Hepatocytes Trigger Growth of Liver Progenitors. Gut 2010, 59, 655–665, doi:10.1136/gut.2009.204354.
- Wynn, T.A. Cellular and Molecular Mechanisms of Fibrosis. J Pathol 2008, 214, 199–210, doi:10.1002/path.2277.
- Tsochatzis, E.A.; Bosch, J.; Burroughs, A.K. Liver Cirrhosis. Lancet 2014, 383, 1749–1761, doi:10.1016/S0140-6736(14)60121-5.
- Zhao, S.-X.; Li, W.-C.; Fu, N.; Kong, L.-B.; Zhang, Q.-S.; Han, F.; Ren, W.-G.; Cui, P.; Du, J.-H.; Wang, B.-Y.; et al. CD14+ Monocytes and CD163+ Macrophages Correlate with the Severity of Liver Fibrosis in Patients with Chronic Hepatitis C. Exp Ther Med 2020, 20, 228, doi:10.3892/etm.2020.9358.
- Douam, F.; Lavillette, D.; Cosset, F.-L. The Mechanism of HCV Entry into Host Cells. Prog Mol Biol Transl Sci 2015, 129, 63–107, doi:10.1016/bs.pmbts.2014.10.003.
- Hemler, M.E. Tetraspanin Functions and Associated Microdomains. Nat. Rev. Mol. Cell Biol. 2005, 6, 801–811, doi:10.1038/nrm1736.
- Berditchevski, F. Complexes of Tetraspanins with Integrins: More than Meets the Eye. J. Cell. Sci. 2001, 114, 4143–4151.
- Alisi, A.; Arciello, M.; Petrini, S.; Conti, B.; Missale, G.; Balsano, C. Focal Adhesion Kinase (FAK) Mediates the Induction of pro-Oncogenic and Fibrogenic Phenotypes in Hepatitis C Virus (HCV)-Infected Cells. PLoS ONE 2012, 7, e44147, doi:10.1371/journal.pone.0044147.
- Vicente-Manzanares, M.; Webb, D.J.; Horwitz, A.R. Cell Migration at a Glance. J. Cell. Sci. 2005, 118, 4917–4919, doi:10.1242/jcs.02662.
- Martínez, S.M.; Crespo, G.; Navasa, M.; Forns, X. Noninvasive Assessment of Liver Fibrosis. Hepatology 2011, 53, 325–335, doi:10.1002/hep.24013.
- Karsdal, M.A.; Manon-Jensen, T.; Genovese, F.; Kristensen, J.H.; Nielsen, M.J.; Sand, J.M.B.; Hansen, N.-U.B.; Bay-Jensen, A.-C.; Bager, C.L.; Krag, A.; et al. Novel Insights into the Function and Dynamics of Extracellular Matrix in Liver Fibrosis. Am. J. Physiol. Gastrointest. Liver Physiol. 2015, 308, G807-830, doi:10.1152/ajpgi.00447.2014.
- Ferrell, L. Liver Pathology: Cirrhosis, Hepatitis, and Primary Liver Tumors. Update and Diagnostic Problems. Mod. Pathol. 2000, 13, 679–704, doi:10.1038/modpathol.3880119.
- Govaere, O.; Cockell, S.; Van Haele, M.; Wouters, J.; Van Delm, W.; Van den Eynde, K.; Bianchi, A.; van Eijsden, R.; Van Steenbergen, W.; Monbaliu, D.; et al. High-Throughput Sequencing Identifies Aetiology-Dependent Differences in Ductular Reaction in Human Chronic Liver Disease. J. Pathol. 2019, 248, 66–76, doi:10.1002/path.5228.
- Trivedi, S.; Murthy, S.; Sharma, H.; Hartlage, A.S.; Kumar, A.; Gadi, S.; Simmonds, P.; Chauhan, L.V.; Scheel, T.K.H.; Billerbeck, E.; et al. Viral Persistence, Liver Disease and Host Response in Hepatitis C-like Virus Rat Model. Hepatology 2017, doi:10.1002/hep.29494.
- Khatun, M.; Ray, R.B. Mechanisms Underlying Hepatitis C Virus-Associated Hepatic Fibrosis. Cells 2019, 8, doi:10.3390/cells8101249.
- Nielsen, M.J.; Karsdal, M.A.; Kazankov, K.; Grønbaek, H.; Krag, A.; Leeming, D.J.; Schuppan, D.; George, J. Fibrosis Is Not Just Fibrosis - Basement Membrane Modelling and Collagen Metabolism Differs between Hepatitis B- and C-Induced Injury. Aliment. Pharmacol. Ther. 2016, 44, 1242–1252, doi:10.1111/apt.13819.
- Guido, M.; Mangia, A.; Faa, G.; Gruppo Italiano Patologi Apparato Digerente (GIPAD); Società Italiana di Anatomia Patologica e Citopatologia Diagnostica/International Academy of Pathology, Italian division (SIAPEC/IAP) Chronic Viral Hepatitis: The Histology Report. Dig Liver Dis 2011, 43 Suppl 4, S331-343, doi:10.1016/S1590-8658(11)60589-6.
- Beltra, J.-C.; Decaluwe, H. Cytokines and Persistent Viral Infections. Cytokine 2016, 82, 4–15, doi:10.1016/j.cyto.2016.02.006.
- Govaere, O.; Petz, M.; Wouters, J.; Vandewynckel, Y.-P.; Scott, E.J.; Topal, B.; Nevens, F.; Verslype, C.; Anstee, Q.M.; Van Vlierberghe, H.; et al. The PDGFRα-Laminin B1-Keratin 19 Cascade Drives Tumor Progression at the Invasive Front of Human Hepatocellular Carcinoma. Oncogene 2017, 36, 6605–6616, doi:10.1038/onc.2017.260.
- Martinez-Quetglas, I.; Pinyol, R.; Dauch, D.; Torrecilla, S.; Tovar, V.; Moeini, A.; Alsinet, C.; Portela, A.; Rodriguez-Carunchio, L.; Solé, M.; et al. IGF2 Is Up-Regulated by Epigenetic Mechanisms in Hepatocellular Carcinomas and Is an Actionable Oncogene Product in Experimental Models. Gastroenterology 2016, 151, 1192–1205, doi:10.1053/j.gastro.2016.09.001.
- Preisser, L.; Miot, C.; Le Guillou-Guillemette, H.; Beaumont, E.; Foucher, E.D.; Garo, E.; Blanchard, S.; Frémaux, I.; Croué, A.; Fouchard, I.; et al. IL-34 and Macrophage Colony-Stimulating Factor Are Overexpressed in Hepatitis C Virus Fibrosis and Induce Profibrotic Macrophages That Promote Collagen Synthesis by Hepatic Stellate Cells. Hepatology 2014, 60, 1879–1890, doi:10.1002/hep.27328.
- Tummala, K.S.; Brandt, M.; Teijeiro, A.; Graña, O.; Schwabe, R.F.; Perna, C.; Djouder, N. Hepatocellular Carcinomas Originate Predominantly from Hepatocytes and Benign Lesions from Hepatic Progenitor Cells. Cell Rep 2017, 19, 584–600, doi:10.1016/j.celrep.2017.03.059.
- Schulze-Krebs, A.; Preimel, D.; Popov, Y.; Bartenschlager, R.; Lohmann, V.; Pinzani, M.; Schuppan, D. Hepatitis C Virus-Replicating Hepatocytes Induce Fibrogenic Activation of Hepatic Stellate Cells. Gastroenterology 2005, 129, 246–258.
- Shin, J.Y.; Hur, W.; Wang, J.S.; Jang, J.W.; Kim, C.W.; Bae, S.H.; Jang, S.K.; Yang, S.-H.; Sung, Y.C.; Kwon, O.-J.; et al. HCV Core Protein Promotes Liver Fibrogenesis via Up-Regulation of CTGF with TGF-Beta1. Exp Mol Med 2005, 37, 138–145, doi:10.1038/emm.2005.19.
- Baiocchini, A.; Montaldo, C.; Conigliaro, A.; Grimaldi, A.; Correani, V.; Mura, F.; Ciccosanti, F.; Rotiroti, N.; Brenna, A.; Montalbano, M.; et al. Extracellular Matrix Molecular Remodeling in Human Liver Fibrosis Evolution. PLoS ONE 2016, 11, e0151736, doi:10.1371/journal.pone.0151736.
- Mormone, E.; Lu, Y.; Ge, X.; Fiel, M.I.; Nieto, N. Fibromodulin, an Oxidative Stress-Sensitive Proteoglycan, Regulates the Fibrogenic Response to Liver Injury in Mice. Gastroenterology 2012, 142, 612-621.e5, doi:10.1053/j.gastro.2011.11.029.
- Benzoubir, N.; Lejamtel, C.; Battaglia, S.; Testoni, B.; Benassi, B.; Gondeau, C.; Perrin-Cocon, L.; Desterke, C.; Thiers, V.; Samuel, D.; et al. HCV Core-Mediated Activation of Latent TGF-β via Thrombospondin Drives the Crosstalk between Hepatocytes and Stromal Environment. J. Hepatol. 2013, 59, 1160–1168, doi:10.1016/j.jhep.2013.07.036.
- Bataller, R.; Paik, Y.; Lindquist, J.N.; Lemasters, J.J.; Brenner, D.A. Hepatitis C Virus Core and Nonstructural Proteins Induce Fibrogenic Effects in Hepatic Stellate Cells. Gastroenterology 2004, 126, 529–540, doi:10.1053/j.gastro.2003.11.018.
- Núñez, O.; Fernández-Martínez, A.; Majano, P.L.; Apolinario, A.; Gómez-Gonzalo, M.; Benedicto, I.; López-Cabrera, M.; Boscá, L.; Clemente, G.; García-Monzón, C.; et al. Increased Intrahepatic Cyclooxygenase 2, Matrix Metalloproteinase 2, and Matrix Metalloproteinase 9 Expression Is Associated with Progressive Liver Disease in Chronic Hepatitis C Virus Infection: Role of Viral Core and NS5A Proteins. Gut 2004, 53, 1665–1672, doi:10.1136/gut.2003.038364.
- Iqbal, J.; Sarkar-Dutta, M.; McRae, S.; Ramachandran, A.; Kumar, B.; Waris, G. Osteopontin Regulates Hepatitis C Virus (HCV) Replication and Assembly by Interacting with HCV Proteins and Lipid Droplets and by Binding to Receptors ΑVβ3 and CD44. J. Virol. 2018, 92, doi:10.1128/JVI.02116-17.
- Shirasaki, T.; Honda, M.; Yamashita, T.; Nio, K.; Shimakami, T.; Shimizu, R.; Nakasyo, S.; Murai, K.; Shirasaki, N.; Okada, H.; et al. The Osteopontin-CD44 Axis in Hepatic Cancer Stem Cells Regulates IFN Signaling and HCV Replication. Scientific Reports 2018, 8, 13143, doi:10.1038/s41598-018-31421-6.
- Jee, M.H.; Hong, K.Y.; Park, J.H.; Lee, J.S.; Kim, H.S.; Lee, S.H.; Jang, S.K. New Mechanism of Hepatic Fibrogenesis: Hepatitis C Virus Infection Induces Transforming Growth Factor Β1 Production through Glucose-Regulated Protein 94. J. Virol. 2015, 90, 3044–3055, doi:10.1128/JVI.02976-15.
- Chida, T.; Ito, M.; Nakashima, K.; Kanegae, Y.; Aoshima, T.; Takabayashi, S.; Kawata, K.; Nakagawa, Y.; Yamamoto, M.; Shimano, H.; et al. Critical Role of CREBH-Mediated Induction of Transforming Growth Factor Β2 by Hepatitis C Virus Infection in Fibrogenic Responses in Hepatic Stellate Cells. Hepatology 2017, 66, 1430–1443, doi:10.1002/hep.29319.
- Kwon, Y.-C.; Sasaki, R.; Meyer, K.; Ray, R. Hepatitis C Virus Core Protein Modulates Endoglin (CD105) Signaling Pathway for Liver Pathogenesis. J. Virol. 2017, 91, doi:10.1128/JVI.01235-17.
- Xue, Y.; Mars, W.M.; Bowen, W.; Singhi, A.D.; Stoops, J.; Michalopoulos, G.K. Hepatitis C Virus Mimics Effects of Glypican-3 on CD81 and Promotes Development of Hepatocellular Carcinomas via Activation of Hippo Pathway in Hepatocytes. Am J Pathol 2018, 188, 1469–1477, doi:10.1016/j.ajpath.2018.02.013.
- Kim, H.; Bose, S.K.; Meyer, K.; Ray, R. Hepatitis C Virus Impairs Natural Killer Cell-Mediated Augmentation of Complement Synthesis. J. Virol. 2014, 88, 2564–2571, doi:10.1128/JVI.02988-13.
- Lu, L.; Zhang, Q.; Wu, K.; Chen, X.; Zheng, Y.; Zhu, C.; Wu, J. Hepatitis C Virus NS3 Protein Enhances Cancer Cell Invasion by Activating Matrix Metalloproteinase-9 and Cyclooxygenase-2 through ERK/P38/NF-ΚB Signal Cascade. Cancer Lett. 2015, 356, 470–478, doi:10.1016/j.canlet.2014.09.027.
- Presser, L.D.; Haskett, A.; Waris, G. Hepatitis C Virus-Induced Furin and Thrombospondin-1 Activate TGF-Β1: Role of TGF-Β1 in HCV Replication. Virology 2011, 412, 284–296, doi:10.1016/j.virol.2010.12.051.
- Sakata, K.; Hara, M.; Terada, T.; Watanabe, N.; Takaya, D.; Yaguchi, S.; Matsumoto, T.; Matsuura, T.; Shirouzu, M.; Yokoyama, S.; et al. HCV NS3 Protease Enhances Liver Fibrosis via Binding to and Activating TGF-β Type I Receptor. Sci Rep 2013, 3, 3243, doi:10.1038/srep03243.
- Wen, C.; He, X.; Ma, H.; Hou, N.; Wei, C.; Song, T.; Zhang, Y.; Sun, L.; Ma, Q.; Zhong, H. Hepatitis C Virus Infection Downregulates the Ligands of the Activating Receptor NKG2D. Cell. Mol. Immunol. 2008, 5, 475–478, doi:10.1038/cmi.2008.60.
- Verga-Gérard, A.; Porcherot, M.; Meyniel-Schicklin, L.; André, P.; Lotteau, V.; Perrin-Cocon, L. Hepatitis C Virus/Human Interactome Identifies SMURF2 and the Viral Protease as Critical Elements for the Control of TGF-β Signaling. FASEB J. 2013, 27, 4027–4040, doi:10.1096/fj.13-229187.
- Li, Y.; Zhang, Q.; Liu, Y.; Luo, Z.; Kang, L.; Qu, J.; Liu, W.; Xia, X.; Liu, Y.; Wu, K.; et al. Hepatitis C Virus Activates Bcl-2 and MMP-2 Expression through Multiple Cellular Signaling Pathways. J. Virol. 2012, 86, 12531–12543, doi:10.1128/JVI.01136-12.
- Chusri, P.; Kumthip, K.; Hong, J.; Zhu, C.; Duan, X.; Jilg, N.; Fusco, D.N.; Brisac, C.; Schaefer, E.A.; Cai, D.; et al. HCV Induces Transforming Growth Factor Β1 through Activation of Endoplasmic Reticulum Stress and the Unfolded Protein Response. Sci Rep 2016, 6, 22487, doi:10.1038/srep22487.
- Choi, S.-H.; Hwang, S.B. Modulation of the Transforming Growth Factor-Beta Signal Transduction Pathway by Hepatitis C Virus Nonstructural 5A Protein. J Biol Chem 2006, 281, 7468–7478, doi:10.1074/jbc.M512438200.
- Naba, A.; Clauser, K.R.; Hoersch, S.; Liu, H.; Carr, S.A.; Hynes, R.O. The Matrisome: In Silico Definition and in Vivo Characterization by Proteomics of Normal and Tumor Extracellular Matrices. Mol. Cell Proteomics 2012, 11, M111.014647, doi:10.1074/mcp.M111.014647.
- Naba, A.; Clauser, K.R.; Whittaker, C.A.; Carr, S.A.; Tanabe, K.K.; Hynes, R.O. Extracellular Matrix Signatures of Human Primary Metastatic Colon Cancers and Their Metastases to Liver. BMC Cancer 2014, 14, 518, doi:10.1186/1471-2407-14-518.
- Decaris, M.L.; Emson, C.L.; Li, K.; Gatmaitan, M.; Luo, F.; Cattin, J.; Nakamura, C.; Holmes, W.E.; Angel, T.E.; Peters, M.G.; et al. Turnover Rates of Hepatic Collagen and Circulating Collagen-Associated Proteins in Humans with Chronic Liver Disease. PloS One 2015, 10, e0123311, doi:10.1371/journal.pone.0123311.
- Asselah, T.; Bièche, I.; Laurendeau, I.; Paradis, V.; Vidaud, D.; Degott, C.; Martinot, M.; Bedossa, P.; Valla, D.; Vidaud, M.; et al. Liver Gene Expression Signature of Mild Fibrosis in Patients with Chronic Hepatitis C. Gastroenterology 2005, 129, 2064–2075, doi:10.1053/j.gastro.2005.09.010.
- Yasui, Y.; Abe, T.; Kurosaki, M.; Matsunaga, K.; Higuchi, M.; Tamaki, N.; Watakabe, K.; Okada, M.; Wang, W.; Shimizu, T.; et al. Non-Invasive Liver Fibrosis Assessment Correlates with Collagen and Elastic Fiber Quantity in Patients with Hepatitis C Virus Infection. Hepatol Res 2019, 49, 33–41, doi:10.1111/hepr.13286.
- Bracht, T.; Schweinsberg, V.; Trippler, M.; Kohl, M.; Ahrens, M.; Padden, J.; Naboulsi, W.; Barkovits, K.; Megger, D.A.; Eisenacher, M.; et al. Analysis of Disease-Associated Protein Expression Using Quantitative Proteomics—Fibulin-5 Is Expressed in Association with Hepatic Fibrosis. J. Proteome Res. 2015, 14, 2278–2286, doi:10.1021/acs.jproteome.5b00053.
- Nielsen, M.J.; Veidal, S.S.; Karsdal, M.A.; Ørsnes-Leeming, D.J.; Vainer, B.; Gardner, S.D.; Hamatake, R.; Goodman, Z.D.; Schuppan, D.; Patel, K. Plasma Pro-C3 (N-Terminal Type III Collagen Propeptide) Predicts Fibrosis Progression in Patients with Chronic Hepatitis C. Liver Int. 2015, 35, 429–437, doi:10.1111/liv.12700.
- Murawaki, Y.; Ikuta, Y.; Koda, M.; Kawasaki, H. Serum Type III Procollagen Peptide, Type IV Collagen 7S Domain, Central Triple-Helix of Type IV Collagen and Tissue Inhibitor of Metalloproteinases in Patients with Chronic Viral Liver Disease: Relationship to Liver Histology. Hepatology 1994, 20, 780–787.
- Valva, P.; Casciato, P.; Diaz Carrasco, J.M.; Gadano, A.; Galdame, O.; Galoppo, M.C.; Mullen, E.; De Matteo, E.; Preciado, M.V. The Role of Serum Biomarkers in Predicting Fibrosis Progression in Pediatric and Adult Hepatitis C Virus Chronic Infection. PLoS One 2011, 6, e23218, doi:10.1371/journal.pone.0023218.
- Lichtinghagen, R.; Michels, D.; Haberkorn, C.I.; Arndt, B.; Bahr, M.; Flemming, P.; Manns, M.P.; Boeker, K.H. Matrix Metalloproteinase (MMP)-2, MMP-7, and Tissue Inhibitor of Metalloproteinase-1 Are Closely Related to the Fibroproliferative Process in the Liver during Chronic Hepatitis C. J. Hepatol. 2001, 34, 239–247, doi:10.1016/s0168-8278(00)00037-4.
- Ljumovic, D.; Diamantis, I.; Alegakis, A.K.; Kouroumalis, E.A. Differential Expression of Matrix Metalloproteinases in Viral and Non-Viral Chronic Liver Diseases. Clin. Chim. Acta 2004, 349, 203–211, doi:10.1016/j.cccn.2004.06.028.
- Martinez-Castillo, M.; Hernandez-Barragan, A.; Flores-Vasconcelos, I.; Galicia-Moreno, M.; Rosique-Oramas, D.; Perez-Hernandez, J.L.; Higuera-De la Tijera, F.; Montalvo-Jave, E.E.; Torre-Delgadillo, A.; Cordero-Perez, P.; et al. Production and Activity of Matrix Metalloproteinases during Liver Fibrosis Progression of Chronic Hepatitis C Patients. World J Hepatol 2021, 13, 218–232, doi:10.4254/wjh.v13.i2.218.
- Murawaki, Y.; Ikuta, Y.; Kawasaki, H. Clinical Usefulness of Serum Tissue Inhibitor of Metalloproteinases (TIMP)-2 Assay in Patients with Chronic Liver Disease in Comparison with Serum TIMP-1. Clin. Chim. Acta 1999, 281, 109–120.
- Dudás, J.; Kovalszky, I.; Gallai, M.; Nagy, J.O.; Schaff, Z.; Knittel, T.; Mehde, M.; Neubauer, K.; Szalay, F.; Ramadori, G. Expression of Decorin, Transforming Growth Factor-Beta 1, Tissue Inhibitor Metalloproteinase 1 and 2, and Type IV Collagenases in Chronic Hepatitis. American Journal of Clinical Pathology 2001, 115, 725–735, doi:10.1309/J8CD-E9C8-X4NG-GTVG.
- Ramnath, D.; Irvine, K.M.; Lukowski, S.W.; Horsfall, L.U.; Loh, Z.; Clouston, A.D.; Patel, P.J.; Fagan, K.J.; Iyer, A.; Lampe, G.; et al. Hepatic Expression Profiling Identifies Steatosis-Independent and Steatosis-Driven Advanced Fibrosis Genes. JCI insight 2018, 3, doi:10.1172/jci.insight.120274.
- Dong, C.; Li, H.-J.; Chang, S.; Liao, H.-J.; Zhang, Z.-P.; Huang, P.; Tang, H.-H. A Disintegrin and Metalloprotease with Thrombospondin Motif 2 May Contribute to Cirrhosis in Humans through the Transforming Growth Factor-β/SMAD Pathway. Gut Liver 2013, 7, 213–220, doi:10.5009/gnl.2013.7.2.213.
- Kuhn, J.; Gressner, O.A.; Götting, C.; Gressner, A.M.; Kleesiek, K. Increased Serum Xylosyltransferase Activity in Patients with Liver Fibrosis. Clin. Chim. Acta 2009, 409, 123–126, doi:10.1016/j.cca.2009.09.013.
- Gressner, O.A.; Gao, C. Monitoring Fibrogenic Progression in the Liver. Clin. Chim. Acta 2014, 433, 111–122, doi:10.1016/j.cca.2014.02.021.
- Zhu, Z.W.; Friess, H.; Wang, L.; Abou-Shady, M.; Zimmermann, A.; Lander, A.D.; Korc, M.; Kleeff, J.; Büchler, M.W. Enhanced Glypican-3 Expression Differentiates the Majority of Hepatocellular Carcinomas from Benign Hepatic Disorders. Gut 2001, 48, 558–564, doi:10.1136/gut.48.4.558.
- Llovet, J.M.; Chen, Y.; Wurmbach, E.; Roayaie, S.; Fiel, M.I.; Schwartz, M.; Thung, S.N.; Khitrov, G.; Zhang, W.; Villanueva, A.; et al. A Molecular Signature to Discriminate Dysplastic Nodules from Early Hepatocellular Carcinoma in HCV Cirrhosis. Gastroenterology 2006, 131, 1758–1767, doi:10.1053/j.gastro.2006.09.014.
- Wurmbach, E.; Chen, Y.; Khitrov, G.; Zhang, W.; Roayaie, S.; Schwartz, M.; Fiel, I.; Thung, S.; Mazzaferro, V.; Bruix, J.; et al. Genome-Wide Molecular Profiles of HCV-Induced Dysplasia and Hepatocellular Carcinoma. Hepatology 2007, 45, 938–947, doi:10.1002/hep.21622.
- Clemente, M.; Núñez, O.; Lorente, R.; Rincón, D.; Matilla, A.; Salcedo, M.; Catalina, M.V.; Ripoll, C.; Iacono, O.L.; Bañares, R.; et al. Increased Intrahepatic and Circulating Levels of Endoglin, a TGF-Beta1 Co-Receptor, in Patients with Chronic Hepatitis C Virus Infection: Relationship to Histological and Serum Markers of Hepatic Fibrosis. J Viral Hepat 2006, 13, 625–632, doi:10.1111/j.1365-2893.2006.00733.x.
- Guéchot, J.; Laudat, A.; Loria, A.; Serfaty, L.; Poupon, R.; Giboudeau, J. Diagnostic Accuracy of Hyaluronan and Type III Procollagen Amino-Terminal Peptide Serum Assays as Markers of Liver Fibrosis in Chronic Viral Hepatitis C Evaluated by ROC Curve Analysis. Clin Chem 1996, 42, 558–563.
- Karsdal, M.A.; Daniels, S.J.; Holm Nielsen, S.; Bager, C.; Rasmussen, D.G.K.; Loomba, R.; Surabattula, R.; Villesen, I.F.; Luo, Y.; Shevell, D.; et al. Collagen Biology and Non-Invasive Biomarkers of Liver Fibrosis. Liver Int. 2020, 40, 736–750, doi:10.1111/liv.14390.
- Taleb, R.S.Z.; Moez, P.; Younan, D.; Eisenacher, M.; Tenbusch, M.; Sitek, B.; Bracht, T. Quantitative Proteome Analysis of Plasma Microparticles for the Characterization of HCV-Induced Hepatic Cirrhosis and Hepatocellular Carcinoma. Proteomics Clin Appl 2017, 11, doi:10.1002/prca.201700014.
- El-Karef, A.; Kaito, M.; Tanaka, H.; Ikeda, K.; Nishioka, T.; Fujita, N.; Inada, H.; Adachi, Y.; Kawada, N.; Nakajima, Y.; et al. Expression of Large Tenascin-C Splice Variants by Hepatic Stellate Cells/Myofibroblasts in Chronic Hepatitis C. J Hepatol 2007, 46, 664–673, doi:10.1016/j.jhep.2006.10.011.
- Benbow, J.H.; Elam, A.D.; Bossi, K.L.; Massengill, D.L.; Brandon-Warner, E.; Anderson, W.E.; Culberson, C.R.; Russo, M.W.; deLemos, A.S.; Schrum, L.W. Analysis of Plasma Tenascin-C in Post-HCV Cirrhosis: A Prospective Study. Dig Dis Sci 2018, 63, 653–664, doi:10.1007/s10620-017-4860-z.
- Choi, S.S.; Claridge, L.C.; Jhaveri, R.; Swiderska-Syn, M.; Clark, P.; Suzuki, A.; Pereira, T.A.; Mi, Z.; Kuo, P.C.; Guy, C.D.; et al. Osteopontin Is Up-Regulated in Chronic Hepatitis C and Is Associated with Cellular Permissiveness for Hepatitis C Virus Replication. Clin Sci (Lond) 2014, 126, 845–855, doi:10.1042/CS20130473.
- Urtasun, R.; Lopategi, A.; George, J.; Leung, T.-M.; Lu, Y.; Wang, X.; Ge, X.; Fiel, M.I.; Nieto, N. Osteopontin, an Oxidant Stress Sensitive Cytokine, up-Regulates Collagen-I via Integrin α(V)β(3) Engagement and PI3K/PAkt/NFκB Signaling. Hepatology 2012, 55, 594–608, doi:10.1002/hep.24701.
- Hackl, N.J.; Bersch, C.; Feick, P.; Antoni, C.; Franke, A.; Singer, M.V.; Nakchbandi, I.A. Circulating Fibronectin Isoforms Predict the Degree of Fibrosis in Chronic Hepatitis C. Scand J Gastroenterol 2010, 45, 349–356, doi:10.3109/00365520903490606.
- Bracht, T.; Mölleken, C.; Ahrens, M.; Poschmann, G.; Schlosser, A.; Eisenacher, M.; Stühler, K.; Meyer, H.E.; Schmiegel, W.H.; Holmskov, U.; et al. Evaluation of the Biomarker Candidate MFAP4 for Non-Invasive Assessment of Hepatic Fibrosis in Hepatitis C Patients. J Transl Med 2016, 14, 1–9, doi:10.1186/s12967-016-0952-3.
- Mölleken, C.; Ahrens, M.; Schlosser, A.; Dietz, J.; Eisenacher, M.; Meyer, H.E.; Schmiegel, W.; Holmskov, U.; Sarrazin, C.; Sorensen, G.L.; et al. Direct-Acting Antivirals-Based Therapy Decreases Hepatic Fibrosis Serum Biomarker Microfibrillar-Associated Protein 4 in Hepatitis C Patients. Clin Mol Hepatol 2018, 25, 42–51, doi:10.3350/cmh.2018.0029.
- Castilla, A.; Prieto, J.; Fausto, N. Transforming Growth Factors Beta 1 and Alpha in Chronic Liver Disease. Effects of Interferon Alfa Therapy. N Engl J Med 1991, 324, 933–940, doi:10.1056/NEJM199104043241401.
- Kinnman, N.; Andersson, U.; Hultcrantz, R. In Situ Expression of Transforming Growth Factor-Beta1-3, Latent Transforming Growth Factor-Beta Binding Protein and Tumor Necrosis Factor-Alpha in Liver Tissue from Patients with Chronic Hepatitis C. Scand J Gastroenterol 2000, 35, 1294–1300, doi:10.1080/003655200453656.
- Divella, R.; Daniele, A.; Gadaleta, C.; Tufaro, A.; Venneri, M.T.; Paradiso, A.; Quaranta, M. Circulating Transforming Growth Factor-β and Epidermal Growth Factor Receptor as Related to Virus Infection in Liver Carcinogenesis. Anticancer Res 2012, 32, 141–145.
- Bader, H.L.; Lambert, E.; Guiraud, A.; Malbouyres, M.; Driever, W.; Koch, M.; Ruggiero, F. Zebrafish Collagen XIV Is Transiently Expressed in Epithelia and Is Required for Proper Function of Certain Basement Membranes. J. Biol. Chem. 2013, 288, 6777–6787, doi:10.1074/jbc.M112.430637.
- Grässel, S.; Unsöld, C.; Schäcke, H.; Bruckner-Tuderman, L.; Bruckner, P. Collagen XVI Is Expressed by Human Dermal Fibroblasts and Keratinocytes and Is Associated with the Microfibrillar Apparatus in the Upper Papillary Dermis. Matrix Biol. 1999, 18, 309–317, doi:10.1016/s0945-053x(99)00019-0.
- Uitto, J.; Pulkkinen, L. Molecular Complexity of the Cutaneous Basement Membrane Zone. Mol. Biol. Rep. 1996, 23, 35–46, doi:10.1007/BF00357071.
- Schuppan, D.; Cramer, T.; Bauer, M.; Strefeld, T.; Hahn, E.G.; Herbst, H. Hepatocytes as a Source of Collagen Type XVIII Endostatin. Lancet 1998, 352, 879–880, doi:10.1016/S0140-6736(05)60006-2.
- Heljasvaara, R.; Aikio, M.; Ruotsalainen, H.; Pihlajaniemi, T. Collagen XVIII in Tissue Homeostasis and Dysregulation - Lessons Learned from Model Organisms and Human Patients. Matrix Biol. 2017, 57–58, 55–75, doi:10.1016/j.matbio.2016.10.002.
- Vadasz, Z.; Kessler, O.; Akiri, G.; Gengrinovitch, S.; Kagan, H.M.; Baruch, Y.; Izhak, O.B.; Neufeld, G. Abnormal Deposition of Collagen around Hepatocytes in Wilson’s Disease Is Associated with Hepatocyte Specific Expression of Lysyl Oxidase and Lysyl Oxidase like Protein-2. J. Hepatol. 2005, 43, 499–507, doi:10.1016/j.jhep.2005.02.052.
- Barry-Hamilton, V.; Spangler, R.; Marshall, D.; McCauley, S.; Rodriguez, H.M.; Oyasu, M.; Mikels, A.; Vaysberg, M.; Ghermazien, H.; Wai, C.; et al. Allosteric Inhibition of Lysyl Oxidase-like-2 Impedes the Development of a Pathologic Microenvironment. Nat. Med. 2010, 16, 1009–1017, doi:10.1038/nm.2208.
- Liu, S.B.; Ikenaga, N.; Peng, Z.-W.; Sverdlov, D.Y.; Greenstein, A.; Smith, V.; Schuppan, D.; Popov, Y. Lysyl Oxidase Activity Contributes to Collagen Stabilization during Liver Fibrosis Progression and Limits Spontaneous Fibrosis Reversal in Mice. FASEB J. 2016, 30, 1599–1609, doi:10.1096/fj.14-268425.
- Ikenaga, N.; Peng, Z.-W.; Vaid, K.A.; Liu, S.B.; Yoshida, S.; Sverdlov, D.Y.; Mikels-Vigdal, A.; Smith, V.; Schuppan, D.; Popov, Y.V. Selective Targeting of Lysyl Oxidase-like 2 (LOXL2) Suppresses Hepatic Fibrosis Progression and Accelerates Its Reversal. Gut 2017, 66, 1697–1708, doi:10.1136/gutjnl-2016-312473.
- Giovannini, C.; Fornari, F.; Indio, V.; Trerè, D.; Renzulli, M.; Vasuri, F.; Cescon, M.; Ravaioli, M.; Perrucci, A.; Astolfi, A.; et al. Direct Antiviral Treatments for Hepatitis C Virus Have Off-Target Effects of Oncologic Relevance in Hepatocellular Carcinoma. Cancers (Basel) 2020, 12, doi:10.3390/cancers12092674.
- Fontana, R.J.; Dienstag, J.L.; Bonkovsky, H.L.; Sterling, R.K.; Naishadham, D.; Goodman, Z.D.; Lok, A.S.F.; Wright, E.C.; Su, G.L.; HALT-C Trial Group Serum Fibrosis Markers Are Associated with Liver Disease Progression in Non-Responder Patients with Chronic Hepatitis C. Gut 2010, 59, 1401–1409, doi:10.1136/gut.2010.207423.
- Medeiros, T.; Saraiva, G.N.; Moraes, L.A.; Gomes, A.C.; Lacerda, G.S.; Leite, P.E.; Esberard, E.B.; Andrade, T.G.; Xavier, A.R.; Quírico-Santos, T.; et al. Liver Fibrosis Improvement in Chronic Hepatitis C after Direct Acting-Antivirals Is Accompanied by Reduced Profibrogenic Biomarkers-a Role for MMP-9/TIMP-1. Dig Liver Dis 2020, doi:10.1016/j.dld.2020.05.004.
- Schwettmann, L.; Wehmeier, M.; Jokovic, D.; Aleksandrova, K.; Brand, K.; Manns, M.P.; Lichtinghagen, R.; Bahr, M.J. Hepatic Expression of A Disintegrin and Metalloproteinase (ADAM) and ADAMs with Thrombospondin Motives (ADAM-TS) Enzymes in Patients with Chronic Liver Diseases. J. Hepatol. 2008, 49, 243–250, doi:10.1016/j.jhep.2008.03.020.
- Bourd-Boittin, K.; Basset, L.; Bonnier, D.; L’helgoualc’h, A.; Samson, M.; Théret, N. CX3CL1/Fractalkine Shedding by Human Hepatic Stellate Cells: Contribution to Chronic Inflammation in the Liver. J. Cell. Mol. Med. 2009, 13, 1526–1535, doi:10.1111/j.1582-4934.2009.00787.x.
- Parkes, J.; Guha, I.N.; Roderick, P.; Harris, S.; Cross, R.; Manos, M.M.; Irving, W.; Zaitoun, A.; Wheatley, M.; Ryder, S.; et al. Enhanced Liver Fibrosis (ELF) Test Accurately Identifies Liver Fibrosis in Patients with Chronic Hepatitis C. J. Viral Hepat. 2011, 18, 23–31, doi:10.1111/j.1365-2893.2009.01263.x.
- Kumar, V.; Kato, N.; Urabe, Y.; Takahashi, A.; Muroyama, R.; Hosono, N.; Otsuka, M.; Tateishi, R.; Omata, M.; Nakagawa, H.; et al. Genome-Wide Association Study Identifies a Susceptibility Locus for HCV-Induced Hepatocellular Carcinoma. Nat. Genet. 2011, 43, 455–458, doi:10.1038/ng.809.
- Zwirner, N.W.; Dole, K.; Stastny, P. Differential Surface Expression of MICA by Endothelial Cells, Fibroblasts, Keratinocytes, and Monocytes. Hum. Immunol. 1999, 60, 323–330, doi:10.1016/s0198-8859(98)00128-1.
- Goto, K.; Kato, N. MICA SNPs and the NKG2D System in Virus-Induced HCC. J. Gastroenterol. 2015, 50, 261–272, doi:10.1007/s00535-014-1000-9.
- Waldhauer, I.; Goehlsdorf, D.; Gieseke, F.; Weinschenk, T.; Wittenbrink, M.; Ludwig, A.; Stevanovic, S.; Rammensee, H.-G.; Steinle, A. Tumor-Associated MICA Is Shed by ADAM Proteases. Cancer Res. 2008, 68, 6368–6376, doi:10.1158/0008-5472.CAN-07-6768.
- Kohga, K.; Takehara, T.; Tatsumi, T.; Ishida, H.; Miyagi, T.; Hosui, A.; Hayashi, N. Sorafenib Inhibits the Shedding of Major Histocompatibility Complex Class I-Related Chain A on Hepatocellular Carcinoma Cells by down-Regulating a Disintegrin and Metalloproteinase 9. Hepatology 2010, 51, 1264–1273, doi:10.1002/hep.23456.
- Kohga, K.; Takehara, T.; Tatsumi, T.; Miyagi, T.; Ishida, H.; Ohkawa, K.; Kanto, T.; Hiramatsu, N.; Hayashi, N. Anticancer Chemotherapy Inhibits MHC Class I-Related Chain a Ectodomain Shedding by Downregulating ADAM10 Expression in Hepatocellular Carcinoma. Cancer Res. 2009, 69, 8050–8057, doi:10.1158/0008-5472.CAN-09-0789.
- Goto, K.; Arai, J.; Stephanou, A.; Kato, N. Novel Therapeutic Features of Disulfiram against Hepatocellular Carcinoma Cells with Inhibitory Effects on a Disintegrin and Metalloproteinase 10. Oncotarget 2018, 9, 18821–18831, doi:10.18632/oncotarget.24568.
- Huang, C.-F.; Huang, C.-Y.; Yeh, M.-L.; Wang, S.-C.; Chen, K.-Y.; Ko, Y.-M.; Lin, C.-C.; Tsai, Y.-S.; Tsai, P.-C.; Lin, Z.-Y.; et al. Genetics Variants and Serum Levels of MHC Class I Chain-Related A in Predicting Hepatocellular Carcinoma Development in Chronic Hepatitis C Patients Post Antiviral Treatment. EBioMedicine 2017, 15, 81–89, doi:10.1016/j.ebiom.2016.11.031.
- Theocharis, A.D.; Skandalis, S.S.; Tzanakakis, G.N.; Karamanos, N.K. Proteoglycans in Health and Disease: Novel Roles for Proteoglycans in Malignancy and Their Pharmacological Targeting. FEBS J. 2010, 277, 3904–3923, doi:10.1111/j.1742-4658.2010.07800.x.
- Shi, Q.; Jiang, J.; Luo, G. Syndecan-1 Serves as the Major Receptor for Attachment of Hepatitis C Virus to the Surfaces of Hepatocytes. J. Virol. 2013, 87, 6866–6875, doi:10.1128/JVI.03475-12.
- Lefèvre, M.; Felmlee, D.J.; Parnot, M.; Baumert, T.F.; Schuster, C. Syndecan 4 Is Involved in Mediating HCV Entry through Interaction with Lipoviral Particle-Associated Apolipoprotein E. PLoS ONE 2014, 9, e95550, doi:10.1371/journal.pone.0095550.
- Matsumoto, A.; Ono, M.; Fujimoto, Y.; Gallo, R.L.; Bernfield, M.; Kohgo, Y. Reduced Expression of Syndecan-1 in Human Hepatocellular Carcinoma with High Metastatic Potential. Int. J. Cancer 1997, 74, 482–491.
- Regős, E.; Karászi, K.; Reszegi, A.; Kiss, A.; Schaff, Z.; Baghy, K.; Kovalszky, I. Syndecan-1 in Liver Diseases. Pathol. Oncol. Res. 2020, 26, 813–819, doi:10.1007/s12253-019-00617-0.
- Pönighaus, C.; Ambrosius, M.; Casanova, J.C.; Prante, C.; Kuhn, J.; Esko, J.D.; Kleesiek, K.; Götting, C. Human Xylosyltransferase II Is Involved in the Biosynthesis of the Uniform Tetrasaccharide Linkage Region in Chondroitin Sulfate and Heparan Sulfate Proteoglycans. J. Biol. Chem. 2007, 282, 5201–5206, doi:10.1074/jbc.M611665200.
- Baghy, K.; Tátrai, P.; Regős, E.; Kovalszky, I. Proteoglycans in Liver Cancer. World Journal of Gastroenterology 2016, 22, 379–393, doi:10.3748/wjg.v22.i1.379.
- Liu, B.; Paranjpe, S.; Bowen, W.C.; Bell, A.W.; Luo, J.-H.; Yu, Y.-P.; Mars, W.M.; Michalopoulos, G.K. Investigation of the Role of Glypican 3 in Liver Regeneration and Hepatocyte Proliferation. Am. J. Pathol. 2009, 175, 717–724, doi:10.2353/ajpath.2009.081129.
- Toretsky, J.A.; Zitomersky, N.L.; Eskenazi, A.E.; Voigt, R.W.; Strauch, E.D.; Sun, C.C.; Huber, R.; Meltzer, S.J.; Schlessinger, D. Glypican-3 Expression in Wilms Tumor and Hepatoblastoma. Journal of Pediatric Hematology/Oncology 2001, 23, 496–499, doi:10.1097/00043426-200111000-00006.
- Bhave, V.S.; Mars, W.; Donthamsetty, S.; Zhang, X.; Tan, L.; Luo, J.; Bowen, W.C.; Michalopoulos, G.K. Regulation of Liver Growth by Glypican 3, CD81, Hedgehog, and Hhex. Am. J. Pathol. 2013, 183, 153–159, doi:10.1016/j.ajpath.2013.03.013.
- Zvibel, I.; Halfon, P.; Fishman, S.; Penaranda, G.; Leshno, M.; Or, A.B.; Halpern, Z.; Oren, R. Syndecan 1 (CD138) Serum Levels: A Novel Biomarker in Predicting Liver Fibrosis Stage in Patients with Hepatitis C. Liver International: Official Journal of the International Association for the Study of the Liver 2009, 29, 208–212, doi:10.1111/j.1478-3231.2008.01830.x.
- Shimizu, Y.; Mizuno, S.; Fujinami, N.; Suzuki, T.; Saito, K.; Konishi, M.; Takahashi, S.; Gotohda, N.; Tada, T.; Toyoda, H.; et al. Plasma and Tumoral Glypican-3 Levels Are Correlated in Patients with Hepatitis C Virus-Related Hepatocellular Carcinoma. Cancer Sci. 2020, 111, 334–342, doi:10.1111/cas.14251.
- Tsuchiya, N.; Sawada, Y.; Endo, I.; Saito, K.; Uemura, Y.; Nakatsura, T. Biomarkers for the Early Diagnosis of Hepatocellular Carcinoma. World Journal of Gastroenterology 2015, 21, 10573–10583, doi:10.3748/wjg.v21.i37.10573.
- Sun, C.K.; Chua, M.-S.; He, J.; So, S.K. Suppression of Glypican 3 Inhibits Growth of Hepatocellular Carcinoma Cells through Up-Regulation of TGF-Β2. Neoplasia 2011, 13, 735–747.
- Regős, E.; Abdelfattah, H.H.; Reszegi, A.; Szilák, L.; Werling, K.; Szabó, G.; Kiss, A.; Schaff, Z.; Kovalszky, I.; Baghy, K. Syndecan-1 Inhibits Early Stages of Liver Fibrogenesis by Interfering with TGFβ1 Action and Upregulating MMP14. Matrix Biol. 2018, 68–69, 474–489, doi:10.1016/j.matbio.2018.02.008.
- Skandalis, S.S.; Karalis, T.T.; Chatzopoulos, A.; Karamanos, N.K. Hyaluronan-CD44 Axis Orchestrates Cancer Stem Cell Functions. Cellular Signalling 2019, 63, 109377, doi:10.1016/j.cellsig.2019.109377.
- Park, N.R.; Cha, J.H.; Jang, J.W.; Bae, S.H.; Jang, B.; Kim, J.-H.; Hur, W.; Choi, J.Y.; Yoon, S.K. Synergistic Effects of CD44 and TGF-Β1 through AKT/GSK-3β/β-Catenin Signaling during Epithelial-Mesenchymal Transition in Liver Cancer Cells. Biochemical and Biophysical Research Communications 2016, 477, 568–574, doi:10.1016/j.bbrc.2016.06.077.
- Castelli, G.; Pelosi, E.; Testa, U. Liver Cancer: Molecular Characterization, Clonal Evolution and Cancer Stem Cells. Cancers 2017, 9, doi:10.3390/cancers9090127.
- Kakehashi, A.; Ishii, N.; Sugihara, E.; Gi, M.; Saya, H.; Wanibuchi, H. CD44 Variant 9 Is a Potential Biomarker of Tumor Initiating Cells Predicting Survival Outcome in Hepatitis C Virus-Positive Patients with Resected Hepatocellular Carcinoma. Cancer Science 2016, 107, 609–618, doi:10.1111/cas.12908.
- Abe, T.; Fukuhara, T.; Wen, X.; Ninomiya, A.; Moriishi, K.; Maehara, Y.; Takeuchi, O.; Kawai, T.; Akira, S.; Matsuura, Y. CD44 Participates in IP-10 Induction in Cells in Which Hepatitis C Virus RNA Is Replicating, through an Interaction with Toll-like Receptor 2 and Hyaluronan. Journal of Virology 2012, 86, 6159–6170, doi:10.1128/JVI.06872-11.
- Lord, M.S.; Tang, F.; Rnjak-Kovacina, J.; Smith, J.G.W.; Melrose, J.; Whitelock, J.M. The Multifaceted Roles of Perlecan in Fibrosis. Matrix Biol 2018, 68–69, 150–166, doi:10.1016/j.matbio.2018.02.013.
- Batmunkh, E.; Tátrai, P.; Szabó, E.; Lódi, C.; Holczbauer, A.; Páska, C.; Kupcsulik, P.; Kiss, A.; Schaff, Z.; Kovalszky, I. Comparison of the Expression of Agrin, a Basement Membrane Heparan Sulfate Proteoglycan, in Cholangiocarcinoma and Hepatocellular Carcinoma. Hum Pathol 2007, 38, 1508–1515, doi:10.1016/j.humpath.2007.02.017.
- Binder, M.J.; McCoombe, S.; Williams, E.D.; McCulloch, D.R.; Ward, A.C. The Extracellular Matrix in Cancer Progression: Role of Hyalectan Proteoglycans and ADAMTS Enzymes. Cancer Letters 2017, 385, 55–64, doi:10.1016/j.canlet.2016.11.001.
- Sobhy, A.; Fakhry M, M.; A Azeem, H.; Ashmawy, A.M.; Omar Khalifa, H. Significance of Biglycan and Osteopontin as Non-Invasive Markers of Liver Fibrosis in Patients with Chronic Hepatitis B Virus and Chronic Hepatitis C Virus. J Investig Med 2019, 67, 681–685, doi:10.1136/jim-2018-000840.
- Roedig, H.; Damiescu, R.; Zeng-Brouwers, J.; Kutija, I.; Trebicka, J.; Wygrecka, M.; Schaefer, L. Danger Matrix Molecules Orchestrate CD14/CD44 Signaling in Cancer Development. Seminars in Cancer Biology 2020, 62, 31–47, doi:10.1016/j.semcancer.2019.07.026.
- Baghy, K.; Iozzo, R.V.; Kovalszky, I. Decorin-TGFβ Axis in Hepatic Fibrosis and Cirrhosis. The Journal of Histochemistry and Cytochemistry: Official Journal of the Histochemistry Society 2012, 60, 262–268, doi:10.1369/0022155412438104.
- Ivanov, A.V.; Bartosch, B.; Smirnova, O.A.; Isaguliants, M.G.; Kochetkov, S.N. HCV and Oxidative Stress in the Liver. Viruses 2013, 5, 439–469, doi:10.3390/v5020439.
- Campello, E.; Radu, C.M.; Zanetto, A.; Bulato, C.; Shalaby, S.; Spiezia, L.; Franceschet, E.; Burra, P.; Russo, F.P.; Simioni, P. Changes in Plasma Circulating Microvesicles in Patients with HCV-Related Cirrhosis after Treatment with Direct-Acting Antivirals. Liver International: Official Journal of the International Association for the Study of the Liver 2020, 40, 913–920, doi:10.1111/liv.14234.
- Gerarduzzi, C.; Hartmann, U.; Leask, A.; Drobetsky, E. The Matrix Revolution: Matricellular Proteins and Restructuring of the Cancer Microenvironment. Cancer Res 2020, 80, 2705–2717, doi:10.1158/0008-5472.CAN-18-2098.
- Ramazani, Y.; Knops, N.; Elmonem, M.A.; Nguyen, T.Q.; Arcolino, F.O.; van den Heuvel, L.; Levtchenko, E.; Kuypers, D.; Goldschmeding, R. Connective Tissue Growth Factor (CTGF) from Basics to Clinics. Matrix Biol 2018, 68–69, 44–66, doi:10.1016/j.matbio.2018.03.007.
- Tanaka, H.; El-Karef, A.; Kaito, M.; Kinoshita, N.; Fujita, N.; Horiike, S.; Watanabe, S.; Yoshida, T.; Adachi, Y. Circulating Level of Large Splice Variants of Tenascin-C Is a Marker of Piecemeal Necrosis Activity in Patients with Chronic Hepatitis C. Liver Int 2006, 26, 311–318, doi:10.1111/j.1478-3231.2005.01229.x.
- Coombes, J.D.; Swiderska-Syn, M.; Dollé, L.; Reid, D.; Eksteen, B.; Claridge, L.; Briones-Orta, M.A.; Shetty, S.; Oo, Y.H.; Riva, A.; et al. Osteopontin Neutralisation Abrogates the Liver Progenitor Cell Response and Fibrogenesis in Mice. Gut 2015, 64, 1120–1131, doi:10.1136/gutjnl-2013-306484.
- Bruha, R.; Vitek, L.; Smid, V. Osteopontin - A Potential Biomarker of Advanced Liver Disease. Ann Hepatol 2020, 19, 344–352, doi:10.1016/j.aohep.2020.01.001.
- Matsue, Y.; Tsutsumi, M.; Hayashi, N.; Saito, T.; Tsuchishima, M.; Toshikuni, N.; Arisawa, T.; George, J. Serum Osteopontin Predicts Degree of Hepatic Fibrosis and Serves as a Biomarker in Patients with Hepatitis C Virus Infection. PLoS One 2015, 10, e0118744, doi:10.1371/journal.pone.0118744.
- Athwal, V.S.; Pritchett, J.; Martin, K.; Llewellyn, J.; Scott, J.; Harvey, E.; Zaitoun, A.M.; Mullan, A.F.; Zeef, L.A.H.; Friedman, S.L.; et al. Publisher Correction: SOX9 Regulated Matrix Proteins Are Increased in Patients Serum and Correlate with Severity of Liver Fibrosis. Sci Rep 2019, 9, 11547, doi:10.1038/s41598-019-47715-2.
- Wang, X.; Lopategi, A.; Ge, X.; Lu, Y.; Kitamura, N.; Urtasun, R.; Leung, T.-M.; Fiel, M.I.; Nieto, N. Osteopontin Induces Ductular Reaction Contributing to Liver Fibrosis. Gut 2014, 63, 1805–1818, doi:10.1136/gutjnl-2013-306373.
- Sia, D.; Villanueva, A.; Friedman, S.L.; Llovet, J.M. Liver Cancer Cell of Origin, Molecular Class, and Effects on Patient Prognosis. Gastroenterology 2017, 152, 745–761, doi:10.1053/j.gastro.2016.11.048.
- Iso, Y.; Sawada, T.; Okada, T.; Kubota, K. Loss of E-Cadherin MRNA and Gain of Osteopontin MRNA Are Useful Markers for Detecting Early Recurrence of HCV-Related Hepatocellular Carcinoma. J Surg Oncol 2005, 92, 304–311, doi:10.1002/jso.20388.
- Mochida, S.; Hashimoto, M.; Matsui, A.; Naito, M.; Inao, M.; Nagoshi, S.; Nagano, M.; Egashira, T.; Mishiro, S.; Fujiwara, K. Genetic Polymorphims in Promoter Region of Osteopontin Gene May Be a Marker Reflecting Hepatitis Activity in Chronic Hepatitis C Patients. Biochem Biophys Res Commun 2004, 313, 1079–1085, doi:10.1016/j.bbrc.2003.12.045.
- Gressner, O.A.; Gressner, A.M. Connective Tissue Growth Factor: A Fibrogenic Master Switch in Fibrotic Liver Diseases. Liver Int 2008, 28, 1065–1079, doi:10.1111/j.1478-3231.2008.01826.x.
- Paradis, V.; Dargere, D.; Vidaud, M.; De Gouville, A.C.; Huet, S.; Martinez, V.; Gauthier, J.M.; Ba, N.; Sobesky, R.; Ratziu, V.; et al. Expression of Connective Tissue Growth Factor in Experimental Rat and Human Liver Fibrosis. Hepatology 1999, 30, 968–976, doi:10.1002/hep.510300425.
- Hora, C.; Negro, F.; Leandro, G.; Oneta, C.M.; Rubbia-Brandt, L.; Muellhaupt, B.; Helbling, B.; Malinverni, R.; Gonvers, J.-J.; Dufour, J.-F.; et al. Connective Tissue Growth Factor, Steatosis and Fibrosis in Patients with Chronic Hepatitis C. Liver Int 2008, 28, 370–376, doi:10.1111/j.1478-3231.2007.01608.x.
- Nagaraja, T.; Chen, L.; Balasubramanian, A.; Groopman, J.E.; Ghoshal, K.; Jacob, S.T.; Leask, A.; Brigstock, D.R.; Anand, A.R.; Ganju, R.K. Activation of the Connective Tissue Growth Factor (CTGF)-Transforming Growth Factor β 1 (TGF-β 1) Axis in Hepatitis C Virus-Expressing Hepatocytes. PLoS One 2012, 7, e46526, doi:10.1371/journal.pone.0046526.
- Williams, M.J.; Clouston, A.D.; Forbes, S.J. Links between Hepatic Fibrosis, Ductular Reaction, and Progenitor Cell Expansion. Gastroenterology 2014, 146, 349–356, doi:10.1053/j.gastro.2013.11.034.
- Vasel, M.; Rutz, R.; Bersch, C.; Feick, P.; Singer, M.V.; Kirschfink, M.; Nakchbandi, I.A. Complement Activation Correlates with Liver Necrosis and Fibrosis in Chronic Hepatitis C. Clin Immunol 2014, 150, 149–156, doi:10.1016/j.clim.2013.11.014.
- Lorenzini, S.; Bird, T.G.; Boulter, L.; Bellamy, C.; Samuel, K.; Aucott, R.; Clayton, E.; Andreone, P.; Bernardi, M.; Golding, M.; et al. Characterisation of a Stereotypical Cellular and Extracellular Adult Liver Progenitor Cell Niche in Rodents and Diseased Human Liver. Gut 2010, 59, 645–654, doi:10.1136/gut.2009.182345.
- Matsumoto, S.; Yamamoto, K.; Nagano, T.; Okamoto, R.; Ibuki, N.; Tagashira, M.; Tsuji, T. Immunohistochemical Study on Phenotypical Changes of Hepatocytes in Liver Disease with Reference to Extracellular Matrix Composition. Liver 1999, 19, 32–38, doi:10.1111/j.1478-3231.1999.tb00006.x.
- Kanta, J.; Dooley, S.; Delvoux, B.; Breuer, S.; D’Amico, T.; Gressner, A.M. Tropoelastin Expression Is Up-Regulated during Activation of Hepatic Stellate Cells and in the Livers of CCl(4)-Cirrhotic Rats. Liver 2002, 22, 220–227.
- Zhu, S.; Ye, L.; Bennett, S.; Xu, H.; He, D.; Xu, J. Molecular Structure and Function of Microfibrillar-Associated Proteins in Skeletal and Metabolic Disorders and Cancers. J Cell Physiol 2020, doi:10.1002/jcp.29893.
- Papke, C.L.; Yanagisawa, H. Fibulin-4 and Fibulin-5 in Elastogenesis and beyond: Insights from Mouse and Human Studies. Matrix Biol 2014, 37, 142–149, doi:10.1016/j.matbio.2014.02.004.
- Kanzaki, T.; Olofsson, A.; Morén, A.; Wernstedt, C.; Hellman, U.; Miyazono, K.; Claesson-Welsh, L.; Heldin, C.H. TGF-Beta 1 Binding Protein: A Component of the Large Latent Complex of TGF-Beta 1 with Multiple Repeat Sequences. Cell 1990, 61, 1051–1061, doi:10.1016/0092-8674(90)90069-q.
- Kusakabe, M.; Cheong, P.-L.; Nikfar, R.; McLennan, I.S.; Koishi, K. The Structure of the TGF-Beta Latency Associated Peptide Region Determines the Ability of the Proprotein Convertase Furin to Cleave TGF-Betas. J Cell Biochem 2008, 103, 311–320, doi:10.1002/jcb.21407.
- Fausto, N.; Mead, J.E.; Gruppuso, P.A.; Castilla, A.; Jakowlew, S.B. Effects of TGF-Beta s in the Liver: Cell Proliferation and Fibrogenesis. Ciba Found. Symp. 1991, 157, 165–174; discussion 174-177.
- Li, G.; Jiang, Q.; Xu, K. CREB Family: A Significant Role in Liver Fibrosis. Biochimie 2019, 163, 94–100, doi:10.1016/j.biochi.2019.05.014.
- Lua, I.; Li, Y.; Zagory, J.A.; Wang, K.S.; French, S.W.; Sévigny, J.; Asahina, K. Characterization of Hepatic Stellate Cells, Portal Fibroblasts, and Mesothelial Cells in Normal and Fibrotic Livers. J. Hepatol. 2016, 64, 1137–1146, doi:10.1016/j.jhep.2016.01.010.
- Kataria, Y.; Deaton, R.J.; Enk, E.; Jin, M.; Petrauskaite, M.; Dong, L.; Goldenberg, J.R.; Cotler, S.J.; Jensen, D.M.; van Breemen, R.B.; et al. Retinoid and Carotenoid Status in Serum and Liver among Patients at High-Risk for Liver Cancer. BMC Gastroenterol 2016, 16, 30, doi:10.1186/s12876-016-0432-5.
- Fabregat, I.; Moreno-Càceres, J.; Sánchez, A.; Dooley, S.; Dewidar, B.; Giannelli, G.; Ten Dijke, P.; IT-LIVER Consortium TGF-β Signalling and Liver Disease. FEBS J 2016, 283, 2219–2232, doi:10.1111/febs.13665.
- Sasaki, R.; Devhare, P.; Ray, R.B.; Ray, R. Hepatitis C Virus-Induced Tumor-Initiating Cancer Stem-like Cells Activate Stromal Fibroblasts in a Xenograft Tumor Model. Hepatology 2017, 66, 1766–1778, doi:10.1002/hep.29346.
- Nelson, D.R.; Gonzalez-Peralta, R.P.; Qian, K.; Xu, Y.; Marousis, C.G.; Davis, G.L.; Lau, J.Y. Transforming Growth Factor-Beta 1 in Chronic Hepatitis C. J Viral Hepat 1997, 4, 29–35, doi:10.1046/j.1365-2893.1997.00124.x.
- Taniguchi, H.; Kato, N.; Otsuka, M.; Goto, T.; Yoshida, H.; Shiratori, Y.; Omata, M. Hepatitis C Virus Core Protein Upregulates Transforming Growth Factor-Beta 1 Transcription. J Med Virol 2004, 72, 52–59, doi:10.1002/jmv.10545.
- Miyanari, Y.; Atsuzawa, K.; Usuda, N.; Watashi, K.; Hishiki, T.; Zayas, M.; Bartenschlager, R.; Wakita, T.; Hijikata, M.; Shimotohno, K. The Lipid Droplet Is an Important Organelle for Hepatitis C Virus Production. Nat. Cell Biol. 2007, 9, 1089–1097, doi:10.1038/ncb1631.
- Waris, G.; Tardif, K.D.; Siddiqui, A. Endoplasmic Reticulum (ER) Stress: Hepatitis C Virus Induces an ER-Nucleus Signal Transduction Pathway and Activates NF-KappaB and STAT-3. Biochem Pharmacol 2002, 64, 1425–1430, doi:10.1016/s0006-2952(02)01300-x.
- Meurer, S.K.; Alsamman, M.; Scholten, D.; Weiskirchen, R. Endoglin in Liver Fibrogenesis: Bridging Basic Science and Clinical Practice. World J Biol Chem 2014, 5, 180–203, doi:10.4331/wjbc.v5.i2.180.
- About, F.; Bibert, S.; Jouanguy, E.; Nalpas, B.; Lorenzo, L.; Rattina, V.; Zarhrate, M.; Hanein, S.; Munteanu, M.; Müllhaupt, B.; et al. Identification of an Endoglin Variant Associated With HCV-Related Liver Fibrosis Progression by Next-Generation Sequencing. Front Genet 2019, 10, 1024, doi:10.3389/fgene.2019.01024.
- Xu, L.; Hui, A.Y.; Albanis, E.; Arthur, M.J.; O’Byrne, S.M.; Blaner, W.S.; Mukherjee, P.; Friedman, S.L.; Eng, F.J. Human Hepatic Stellate Cell Lines, LX-1 and LX-2: New Tools for Analysis of Hepatic Fibrosis. Gut 2005, 54, 142–151, doi:10.1136/gut.2004.042127.
- Florimond, A.; Chouteau, P.; Bruscella, P.; Le Seyec, J.; Mérour, E.; Ahnou, N.; Mallat, A.; Lotersztajn, S.; Pawlotsky, J.-M. Human Hepatic Stellate Cells Are Not Permissive for Hepatitis C Virus Entry and Replication. Gut 2015, 64, 957–965, doi:10.1136/gutjnl-2013-305634.
- Aoudjehane, L.; Bisch, G.; Scatton, O.; Granier, C.; Gaston, J.; Housset, C.; Roingeard, P.; Cosset, F.-L.; Perdigao, F.; Balladur, P.; et al. Infection of Human Liver Myofibroblasts by Hepatitis C Virus: A Direct Mechanism of Liver Fibrosis in Hepatitis C. PLoS ONE 2015, 10, e0134141, doi:10.1371/journal.pone.0134141.
- Devhare, P.B.; Sasaki, R.; Shrivastava, S.; Di Bisceglie, A.M.; Ray, R.; Ray, R.B. Exosome-Mediated Intercellular Communication between Hepatitis C Virus-Infected Hepatocytes and Hepatic Stellate Cells. J Virol 2017, 91, doi:10.1128/JVI.02225-16.
- Bartosch, B. Piecing Together the Key Players of Fibrosis in Chronic Hepatitis C: What Roles Do Non-Hepatic Liver Resident Cell Types Play? Gut 2015, 64, 862–863, doi:10.1136/gutjnl-2014-307957.
- Foschi, F.G.; Domenicali, M.; Giacomoni, P.; Dall’Aglio, A.C.; Conti, F.; Borghi, A.; Bevilacqua, V.; Napoli, L.; Mirici, F.; Cucchetti, A.; et al. Is There an Association between Commonly Employed Biomarkers of Liver Fibrosis and Liver Stiffness in the General Population? Ann Hepatol 2020, 19, 380–387, doi:10.1016/j.aohep.2020.04.003.
- Meissner, E.G.; McLaughlin, M.; Matthews, L.; Gharib, A.M.; Wood, B.J.; Levy, E.; Sinkus, R.; Virtaneva, K.; Sturdevant, D.; Martens, C.; et al. Simtuzumab Treatment of Advanced Liver Fibrosis in HIV and HCV-Infected Adults: Results of a 6-Month Open-Label Safety Trial. Liver Int. 2016, 36, 1783–1792, doi:10.1111/liv.13177.
- Roehlen, N.; Crouchet, E.; Baumert, T.F. Liver Fibrosis: Mechanistic Concepts and Therapeutic Perspectives. Cells 2020, 9, doi:10.3390/cells9040875.
This entry is adapted from the peer-reviewed paper 10.3390/cancers13092270