 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Anthony Bouter | + 1809 word(s) | 1809 | 2021-05-26 05:34:11 | | | |
| 2 | Nora Tang | Meta information modification | 1809 | 2021-05-31 02:54:30 | | |
Video Upload Options
Muscular dystrophies constitute a group of genetic disorders that cause weakness and progressive loss of skeletal muscle mass. Among them, Miyoshi muscular dystrophy 1 (MMD1), limb girdle muscular dystrophy type R2 (LGMDR2/2B), and LGMDR12 (2L) are characterized by mutation in gene encoding key membrane-repair protein, which leads to severe dysfunctions in sarcolemma repair. Cell membrane disruption is a physiological event induced by mechanical stress, such as muscle contraction and stretching. Like many eukaryotic cells, muscle fibers possess a protein machinery ensuring fast resealing of damaged plasma membrane. Members of the annexins A (ANXA) family belong to this protein machinery. ANXA are small soluble proteins, twelve in number in humans, which share the property of binding to membranes exposing negatively-charged phospholipids in the presence of calcium (Ca2+). Many ANXA have been reported to participate in membrane repair of varied cell types and species, including human skeletal muscle cells in which they may play a collective role in protection and repair of the sarcolemma. Here, we discuss the participation of ANXA in membrane repair of healthy skeletal muscle cells and how dysregulation of ANXA expression may impact the clinical severity of muscular dystrophies.
1. Introduction
Membrane ruptures induced by mechanical stress, such as contraction, stretching, or shearing, compromise cellular homeostasis and lead to cell death in the absence of fast resealing [1][2]. Frequency of cell membrane disruption is high in mammal tissues subjected to severe mechanical constraints, such as cardiac or skeletal muscle, epithelia, and endothelium [3][4][5][6]. For instance, adult rat muscle exposed to eccentric contractions exhibits about 20% of damaged myofibers, instead of 3.1% in basal conditions [5]. Healthy myofibers are able to repair disruptions of the sarcolemma (cell membrane of muscle cell) and survive: the rate of damaged myofibers dropped indeed to 5.6%, 24 h post-exercise [5]. How is skeletal muscle cell able to repair a damaged sarcolemma? This issue continues to be widely debated and the subject of intense research, even if the protein machinery starts being identified (see below). Discrepancies in experimental data have been indeed observed, which may result from the existence of different repair mechanisms depending on the cell type and the damage nature. While a membrane lesion less than 0.2 μm can be repaired passively, tension exerted by actin cytoskeleton on plasma membrane induces larger lesions and requires the intervention of active repair mechanisms [1][2]. These mechanisms are mostly triggered by the influx of extracellular calcium (Ca2+), which rushes into the cell through the membrane breach and invade the cytoplasm. The gradient of Ca2+ concentration created within the cytoplasm activates proteins of the membrane repair machinery, which then move towards the rupture site [7].
The absence of membrane repair causes the death of damaged cells and may contribute to tissue degeneration and the development of degenerative diseases [1]. A defective membrane repair machinery is observed in MMD1 [8][9], LGMDR1 (formerly 2A) [10], LGMDR2 (2B) [8], LGMD1C [11], and LGMDR12 (2L) (see chapter 4 for the nomenclature of LGMD) [12][13]. In addition, in certain forms of muscular dystrophy, such as Duchenne muscular dystrophy (DMD), the frequency of sarcolemma disruption is far higher than in normal muscle, which may lead to wear of the membrane repair machinery [5][14]. Here, we discuss the interplay between muscular dystrophies and sarcolemma repair and how dysregulations in ANXA expression may impact the severity of the disease.
2. Anatomy of Skeletal Muscle
Human body is made up of approximately 650 muscles, with variability between individuals. Skeletal muscle tissue is itself made up of a collection of cell populations bounded by an envelope of fibrous connective tissue, called epimysium (Figure 1) [15]. This envelope ensures the maintenance and protection of the muscle during contraction, and is also the link between muscle and bones. A muscle is subdivided into a set of muscle bundles which are made up of several dozen muscle cells, all of which are surrounded by a connective sheath, called perimysium (Figure 1). Perimysium helps to structure the muscle and also to anchor muscle to bones at the level of tendons. The basal lamina or endomysium constitutes an extracellular matrix sheath that surrounds each muscle cell within a bundle (Figure 1). This extracellular matrix, essentially composed of collagen, ensures the stability of the muscle fiber by interaction with the intracellular cytoskeleton. It also ensures the cohesion of muscle bundle by connecting neighboring muscle cells.
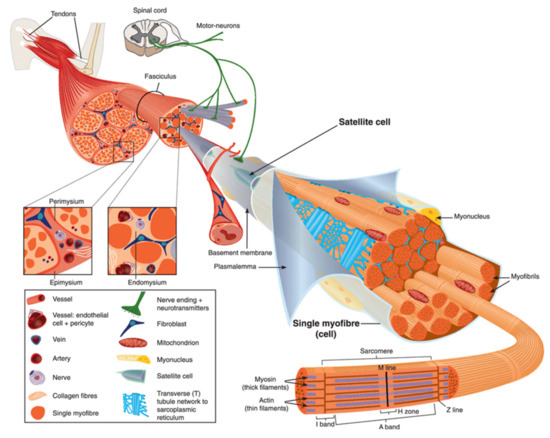
Figure 1. Scheme of skeletal muscle and associated structures. (Left-hand part) Epimysium, perimysium, and endomysium constitute three connective tissue layers that form the lattice network and associated basement membranes in which myofibers regenerate after injury. The epimysium is the outer layer that surrounds the entire muscle and is contiguous with the tendon and endosteum (fascia surrounding bone). The perimysium surrounds bundles of myofibers. The endomysium is located between individual muscle fibers. (Right-hand part) Satellite cells are located between basement membrane and sarcolemma. Sarcolemma bounds each myofiber, which is composed by multiple nuclei and the sarcoplasm that contains mitochondria, sarcoplasmic reticulum and myofibrils. The myofibril is the contractile unit of a myofiber. Specialized cytoskeleton within the myofibril forms repeated structures, called sarcomeres, which appear as a succession of light and dark bands under polarized light optical microscopy. The sarcoplasmic reticulum is the major provider of Ca2+ required for muscle contraction. It is connected to transverse tubules that surround sarcomeres. Adapted from Reference [15] with the Permission 5036470714502 from John Wiley and sons.
Skeletal muscle is made up of two main cell populations: skeletal muscle cells, also called muscle fibers or myofibers or myocytes, and mononuclear stem cells, called satellite cells (Figure 1). Satellite cells are generally quiescent, with the ability to proliferate, differentiate and fuse to form new myofibers. These cells are considered to be the main contributor to post-natal muscle growth and maintenance [16].
Myofibers are definitely special cells in terms of morphology and function. They exhibit a tubular and elongated shape and measure from a few hundred µm to several tens of centimeters in length for a diameter between 10 and 100 µm. Sarcolemma defines cytoplasm (sarcoplasm), which is composed by mitochondria, specialized endoplasmic (sarcoplasmic) reticulum, as well as a set of basic organelles, and a large number of nuclei located on the periphery of the cell.
Sarcolemma is subjected to severe mechanical stress, more than in any other cell type, due to its huge surface and to contraction and stretching processes that it regularly undergoes [17]. To cope with these mechanical stresses, sarcolemma is supported by an exceptional protein framework, composed in particular of dystrophin, dystroglycans, and sarcoglycans [18]. Unrepaired sarcolemma damage leads to the death of myofiber, which is followed by inflammation, especially through the infiltration of macrophages, and regeneration phases, during which the satellite cells proliferate, differentiate and fuse to form a new fiber [19]. Satellite cells are not the only helping cells in skeletal muscle repair. Macrophages have been also reported to mediate sarcolemma repair through a mechanism involving dysferlin (DYSF) and phosphatidylserine (PS) [20]. DYSF accumulated at the membrane disruption site, subsequently to influx of Ca2+, promotes the accumulation of PS in the outer leaflet of the sarcolemma, which triggers the recruitment of macrophages. Excess of membrane and protein used during the resealing process and accumulated at the disruption site is engulfed by macrophages, which enables cell membrane integrity to be regained [20].
3. ANXA and Muscular Dystrophies
In 2003, Bansal and collaborators revealed for the first time a direct link between a failure in membrane repair, caused by mutations in DYSF gene, and the development of a muscular dystrophy, e.g., LGMDR2 (2B) [8]. The fundamental role of ANXA in membrane repair questions their implication in the development of muscular dystrophies. In humans, no correlation has been made to date between muscular dystrophy and alteration of an ANXA gene. However, for the same genetic mutation, patients suffering from muscular dystrophy may exhibit significant differences in clinical signs. Symptoms may vary in nature, as well as in severity [21][22][23][24]. These observations led to the hypothesis that genetic modifiers may exist in muscular dystrophies, including ANXA.
ANXA1 and ANXA2 interact with DYSF to mediate sarcolemma repair [25] and both ANXA have been reported to be upregulated in Italian [26], American [27], or Australian patients [28] suffering from dysferlinopathies. Overexpression of these ANXA is likely an attempt to counteract the absence of DYSF and restore cell membrane repair ability. It has been revealed that excess of ANXA2 that leaks from injured myofibers activates muscle-resident fibro/adipogenic precursors that differentiate into adipocytes, which gradually replace dysferlin-deficient myofibers leading to muscle degeneration [29]. ANXA2 may act, therefore, as a modifying factor which strongly influences, in a negative way, clinical consequences of dysferlinopathies. Overexpression of ANXA2 is also observed in DMD, Becker muscular dystrophy or LMGDR12 and shedding of ANX-positive vesicles have been shown in ANO5-knockout myofibers (LMGDR12), suggesting these diseases may result from fibrotic or adipogenic replacement of myofibers [30][26]. Recently, an increase of 32% in the expression of ANXA2 has been also observed in a rat model of desminopathy [31].
ANXA1 and ANXA2 are susceptible to cleavage by calpains [32], which may be critical for their function in membrane repair [25][33][34]. In calpainopathies, such as LGMDR1 (2A) [10], therefore, it is expected that calpains deficiency may lead to misfunction of ANXA and impairment of membrane resealing.
In addition, a loss of function of ANXA1 and ANXA6 is observed in LGMDR12 and DMD. In damaged ANO5-knockout myofibers (LGMDR12), accumulation of both ANXA is reduced, altering the tight repair cap structure [30]. In DMD, ANXA1 and ANXA6 present a reduced expression leading to exacerbated sarcolemmal injury and delayed repair cap formation due to overexpression of osteopontin [24].
The role played by ANXA6 as a genetic modifier of muscular dystrophies is definitely the most described. It has been reported that ANXA6 knockdown in a zebrafish model of dysferlinopathy reinforces the dystrophic phenotype [35]. In addition, a truncated form of ANXA6, named ANXA6N32, has been identified in Sgcg-null mouse, a model of LGMDR5 (2C) [36] and in dysferlinopathic mice [37]. ANXA6N32 dramatically impairs translocation of the full-length ANXA6 to the membrane disruption site, disrupts the protein scaffold that is pivotal for membrane resealing, and enhances muscular dystrophy [36][37].
Finally, ANXA7 has been also reported as disturbed in skeletal muscle from patients suffering from DMD and MDX mouse, whereas normal muscle contains specifically a 51-kDa ANXA7 isoform, dystrophic muscle exhibits the additional 47-kDa isoform, usually found in undifferentiated myoblasts [38][39]. During progression of the disease, ANXA7 is gradually retrieved in higher concentration in the serum of patients, suggesting the absence of membrane resealing of injured myofibers and the leak of ANXA7 [38]. If its participation in sarcolemma repair remains to be established, ANXA7 has been shown to mediate membrane repair in cancer cells by enabling assembly of the ESCRT-III complex [40].
Understanding how ANXA can modify the evolution of muscular dystrophies remains a huge project. In particular, most hitherto carried-out studies have used animal models and some differences may exist in the etiology and severity of muscular dystrophies between humans and animals. It will be, therefore, interesting in the near future to be able to explore these questions in human skeletal muscle cells.
References
- Dias, C.; Nylandsted, J. Plasma membrane integrity in health and disease: Significance and therapeutic potential. Cell Discov. 2021, 7, 4.
- Zhen, Y.; Radulovic, M.; Vietri, M.; Stenmark, H. Sealing holes in cellular membranes. EMBO J. 2021, 40, e106922.
- McNeil, P.L.; Ito, S. Gastrointestinal cell plasma membrane wounding and resealing in vivo. Gastroenterology 1989, 96, 1238–1248.
- Yu, Q.C.; McNeil, P.L. Transient disruptions of aortic endothelial cell plasma membranes. Am. J. Pathol. 1992, 141, 1349–1360.
- McNeil, P.L.; Khakee, R. Disruptions of muscle fiber plasma membranes. Role in exercise-induced damage. Am. J. Pathol. 1992, 140, 1097–1109.
- Clarke, M.S.; Caldwell, R.W.; Chiao, H.; Miyake, K.; McNeil, P.L. Contraction-induced cell wounding and release of fibroblast growth factor in heart. Circ. Res. 1995, 76, 927–934.
- Horn, A.; Jaiswal, J.K. Cellular mechanisms and signals that coordinate plasma membrane repair. Cell. Mol. Life Sci. 2018, 75, 3751–3770.
- Bansal, D.; Miyake, K.; Vogel, S.S.; Groh, S.; Chen, C.-C.; Williamson, R.; McNeil, P.L.; Campbell, K.P. Defective membrane repair in dysferlin-deficient muscular dystrophy. Nature 2003, 423, 168–172.
- Liu, J.; Aoki, M.; Illa, I.; Wu, C.; Fardeau, M.; Angelini, C.; Serrano, C.; Andoni Urtizberea, J.; Hentati, F.; Hamida, M.B.; et al. Dysferlin, a novel skeletal muscle gene, is mutated in Miyoshi myopathy and limb girdle muscular dystrophy. Nat. Genet. 1998, 20, 31–36.
- Richard, I.; Broux, O.; Allamand, V.; Fougerousse, F.; Chiannilkulchai, N.; Bourg, N.; Brenguier, L.; Devaud, C.; Pasturaud, P.; Roudaut, C.; et al. Mutations in the proteolytic enzyme calpain 3 cause limb-girdle muscular dystrophy type 2A. Cell 1995, 81, 27–40.
- Minetti, C.; Sotgia, F.; Bruno, C.; Scartezzini, P.; Broda, P.; Bado, M.; Masetti, E.; Mazzocco, M.; Egeo, A.; Donati, M.A.; et al. Mutations in the caveolin-3 gene cause autosomal dominant limb-girdle muscular dystrophy. Nat. Genet. 1998, 18, 365–368.
- Griffin, D.A.; Johnson, R.W.; Whitlock, J.M.; Pozsgai, E.R.; Heller, K.N.; Grose, W.E.; Arnold, W.D.; Sahenk, Z.; Hartzell, H.C.; Rodino-Klapac, L.R. Defective membrane fusion and repair in Anoctamin5 -deficient muscular dystrophy. Hum. Mol. Genet. 2016, 25, 1900–1911.
- Xu, J.; Xu, L.; Lau, Y.S.; Gao, Y.; Moore, S.A.; Han, R. A novel ANO5 splicing variant in a LGMD2L patient leads to production of a truncated aggregation-prone Ano5 peptide. J. Pathol. Clin. Res. 2018, 4, 135–145.
- Clarke, M.S.F.; Khakee, R.; McNeil, P.L. Loss of cytoplasmic basic fibroblast growth factor from physiologically wounded myofibers of normal and dystrophic muscle. J. Cell Sci. 1993, 106, 121–133.
- Tajbakhsh, S. Skeletal muscle stem cells in developmental versus regenerative myogenesis. J. Intern. Med. 2009, 266, 372–389.
- Wang, Y.X.; Rudnicki, M.A. Satellite cells, the engines of muscle repair. Nat. Rev. Mol. Cell Biol. 2012, 13, 127–133.
- McNeil, P.L.; Steinhardt, R.A. Plasma membrane disruption: Repair, prevention, adaptation. Ann. Rev. Cell Dev. Biol. 2003, 19, 697–731.
- Davies, K.E.; Nowak, K.J. Molecular mechanisms of muscular dystrophies: Old and new players. Nat. Rev. Mol. Cell Biol. 2006, 7, 762–773.
- Abmayr, S.M.; Pavlath, G.K. Myoblast fusion: Lessons from flies and mice. Development 2012, 139, 641–656.
- Middel, V.; Zhou, L.; Takamiya, M.; Beil, T.; Shahid, M.; Roostalu, U.; Grabher, C.; Rastegar, S.; Reischl, M.; Nienhaus, G.U.; et al. Dysferlin-mediated phosphatidylserine sorting engages macrophages in sarcolemma repair. Nat. Commun. 2016, 7, 12875.
- Fee, D.B.; So, Y.T.; Barraza, C.; Figueroa, K.P.; Pulst, S.M. Phenotypic variability associated with Arg26Gln mutation in caveolin3. Muscle Nerve 2004, 30, 375–378.
- Vilchez, J.J.; Gallano, P.; Gallardo, E.; Lasa, A.; Rojas-García, R.; Freixas, A.; De Luna, N.; Calafell, F.; Sevilla, T.; Mayordomo, F.; et al. Identification of a novel founder mutation in the DYSF gene causing clinical variability in the spanish population. Arch. Neurol. 2005, 62, 1256–1259.
- Heydemann, A.; Huber, J.M.; Demonbreun, A.; Hadhazy, M.; McNally, E.M. Genetic background influences muscular dystrophy. Neuromuscul. Disord. 2005, 15, 601–609.
- Quattrocelli, M.; Capote, J.; Ohiri, J.C.; Warner, J.L.; Vo, A.H.; Earley, J.U.; Hadhazy, M.; Demonbreun, A.R.; Spencer, M.J.; McNally, E.M. Genetic modifiers of muscular dystrophy act on sarcolemmal resealing and recovery from injury. PLoS Genet. 2017, 13, e1007070.
- Lennon, N.J.; Kho, A.; Bacskai, B.J.; Perlmutter, S.L.; Hyman, B.T.; Brown, R.H. Dysferlin interacts with annexins A1 and A2 and mediates sarcolemmal wound-healing. J. Biol. Chem. 2003, 278, 50466–50473.
- Cagliani, R.; Magri, F.; Toscano, A.; Merlini, L.; Fortunato, F.; Lamperti, C.; Rodolico, C.; Prelle, A.; Sironi, M.; Aguennouz, M.; et al. Mutation finding in patients with dysferlin deficiency and role of the dysferlin interacting proteins annexin A1 and A2 in muscular dystrophies. Hum. Mutat. 2005, 26, 283.
- Kesari, A.; Fukuda, M.; Knoblach, S.; Bashir, R.; Nader, G.A.; Rao, D.; Nagaraju, K.; Hoffman, E.P. Dysferlin deficiency shows compensatory induction of Rab27A/Slp2a that may contribute to inflammatory onset. Am. J. Pathol. 2008, 173, 1476–1487.
- Waddell, L.B.; Lemckert, F.A.; Zheng, X.F.; Tran, J.; Evesson, F.J.; Hawkes, J.M.; Lek, A.; Street, N.E.; Lin, P.; Clarke, N.F.; et al. Dysferlin, annexin A1, and mitsugumin 53 are upregulated in muscular dystrophy and localize to longitudinal tubules of the T-system with stretch. J. Neuropathol. Exp. Neurol. 2011, 70, 302–313.
- Hogarth, M.W.; Defour, A.; Lazarski, C.; Gallardo, E.; Manera, J.D.; Partridge, T.A.; Nagaraju, K.; Jaiswal, J.K. Fibroadipogenic progenitors are responsible for muscle loss in limb girdle muscular dystrophy 2B. Nat. Commun. 2019, 10, 2430.
- Foltz, S.J.; Cui, Y.Y.; Choo, H.J.; Hartzell, H.C. ANO5 ensures trafficking of annexins in wounded myofibers. J. Cell Biol. 2021, 220, e202007059.
- Langer, H.T.; Mossakowski, A.A.; Willis, B.J.; Grimsrud, K.N.; Wood, J.A.; Lloyd, K.C.K.; Zbinden-Foncea, H.; Baar, K. Generation of desminopathy in rats using CRISPR-Cas9. J. Cachexia. Sarcopenia Muscle 2020, 11, 1364–1376.
- Babiychuk, E.B.; Monastyrskaya, K.; Burkhard, F.C.; Wray, S.; Draeger, A. Modulating signaling events in smooth muscle: Cleavage of annexin 2 abolishes its binding to lipid rafts. FASEB J. 2002, 16, 1177–1184.
- Mellgren, R.L.; Miyake, K.; Kramerova, I.; Spencer, M.J.; Bourg, N.; Bartoli, M.; Richard, I.; Greer, P.A.; McNeil, P.L. Calcium-dependent plasma membrane repair requires m- or μ-calpain, but not calpain-3, the proteasome, or caspases. Biochim. Biophys. Acta Mol. Cell Res. 2009, 1793, 1886–1893.
- Mellgren, R.L.; Huang, X. Fetuin A stabilizes m-calpain and facilitates plasma membrane repair. J. Biol. Chem. 2007, 282, 35868–35877.
- Roostalu, U.; Strähle, U. In vivo imaging of molecular interactions at damaged sarcolemma. Dev. Cell 2012, 22, 515–529.
- Swaggart, K.A.; Demonbreun, A.R.; Vo, A.H.; Swanson, K.E.; Kim, E.Y.; Fahrenbach, J.P.; Holley-Cuthrell, J.; Eskin, A.; Chen, Z.; Squire, K.; et al. Annexin A6 modifies muscular dystrophy by mediating sarcolemmal repair. Proc. Natl. Acad. Sci. USA 2014, 111, 6004–6009.
- Demonbreun, A.R.; Allen, M.V.; Warner, J.L.; Barefield, D.Y.; Krishnan, S.; Swanson, K.E.; Earley, J.U.; McNally, E.M. Enhanced muscular dystrophy from loss of dysferlin is accompanied by impaired annexin A6 translocation after sarcolemmal disruption. Am. J. Pathol. 2016, 186, 1610–1622.
- Selbert, S.; Fischer, P.; Menke, A.; Jockusch, H.; Pongratz, D.; Noegel, A.A. Annexin VII relocalization as a result of dystrophin deficiency. Exp. Cell Res. 1996, 222, 199–208.
- Selbert, S.; Fischer, P.; Pongratz, D.; Stewart, M.; Noegel, A.A. Expression and localization of annexin VII (synexin) in muscle cells. J. Cell Sci. 1995, 108, 85–95.
- Sønder, S.L.; Boye, T.L.; Tölle, R.; Dengjel, J.; Maeda, K.; Jäättelä, M.; Simonsen, A.C.; Jaiswal, J.K.; Nylandsted, J. Annexin A7 is required for ESCRT III-mediated plasma membrane repair. Sci. Rep. 2019, 9, 6726.


