Muscular dystrophies constitute a group of genetic disorders that cause weakness and progressive loss of skeletal muscle mass. Among them, Miyoshi muscular dystrophy 1 (MMD1), limb girdle muscular dystrophy type R2 (LGMDR2/2B), and LGMDR12 (2L) are characterized by mutation in gene encoding key membrane-repair protein, which leads to severe dysfunctions in sarcolemma repair. Cell membrane disruption is a physiological event induced by mechanical stress, such as muscle contraction and stretching. Like many eukaryotic cells, muscle fibers possess a protein machinery ensuring fast resealing of damaged plasma membrane. Members of the annexins A (ANXA) family belong to this protein machinery. ANXA are small soluble proteins, twelve in number in humans, which share the property of binding to membranes exposing negatively-charged phospholipids in the presence of calcium (Ca2+). Many ANXA have been reported to participate in membrane repair of varied cell types and species, including human skeletal muscle cells in which they may play a collective role in protection and repair of the sarcolemma. Here, we discuss the participation of ANXA in membrane repair of healthy skeletal muscle cells and how dysregulation of ANXA expression may impact the clinical severity of muscular dystrophies.
- annexins
- muscular dystrophy
- membrane repair
- skeletal muscle
- genetic modifiers
- LGMD
- FSHD
- DMD
1. Introduction
Membrane ruptures induced by mechanical stress, such as contraction, stretching, or shearing, compromise cellular homeostasis and lead to cell death in the absence of fast resealing [1][2]. Frequency of cell membrane disruption is high in mammal tissues subjected to severe mechanical constraints, such as cardiac or skeletal muscle, epithelia, and endothelium [3][4][5][6]. For instance, adult rat muscle exposed to eccentric contractions exhibits about 20% of damaged myofibers, instead of 3.1% in basal conditions [5]. Healthy myofibers are able to repair disruptions of the sarcolemma (cell membrane of muscle cell) and survive: the rate of damaged myofibers dropped indeed to 5.6%, 24 h post-exercise [5]. How is skeletal muscle cell able to repair a damaged sarcolemma? This issue continues to be widely debated and the subject of intense research, even if the protein machinery starts being identified (see below). Discrepancies in experimental data have been indeed observed, which may result from the existence of different repair mechanisms depending on the cell type and the damage nature. While a membrane lesion less than 0.2 μm can be repaired passively, tension exerted by actin cytoskeleton on plasma membrane induces larger lesions and requires the intervention of active repair mechanisms [1][2]. These mechanisms are mostly triggered by the influx of extracellular calcium (Ca
2+
2+
The absence of membrane repair causes the death of damaged cells and may contribute to tissue degeneration and the development of degenerative diseases [1]. A defective membrane repair machinery is observed in MMD1 [8][9], LGMDR1 (formerly 2A) [10], LGMDR2 (2B) [8], LGMD1C [11], and LGMDR12 (2L) (see chapter 4 for the nomenclature of LGMD) [12][13]. In addition, in certain forms of muscular dystrophy, such as Duchenne muscular dystrophy (DMD), the frequency of sarcolemma disruption is far higher than in normal muscle, which may lead to wear of the membrane repair machinery [5][14]. Here, we discuss the interplay between muscular dystrophies and sarcolemma repair and how dysregulations in ANXA expression may impact the severity of the disease.
2. Anatomy of Skeletal Muscle
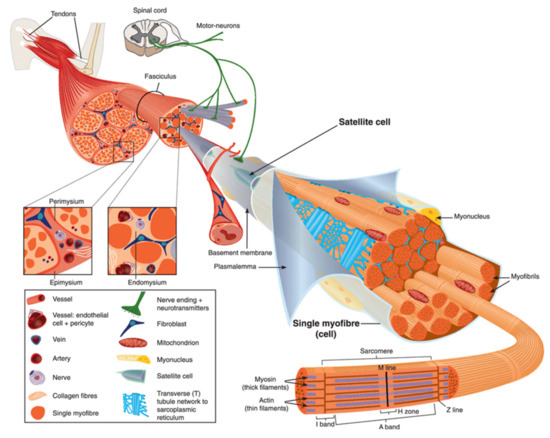
Figure 1.
Left-hand part
Right-hand part
2+
2+
3. ANXA and Muscular Dystrophies
In 2003, Bansal and collaborators revealed for the first time a direct link between a failure in membrane repair, caused by mutations in DYSF gene, and the development of a muscular dystrophy, e.g., LGMDR2 (2B) [8]. The fundamental role of ANXA in membrane repair questions their implication in the development of muscular dystrophies. In humans, no correlation has been made to date between muscular dystrophy and alteration of an ANXA gene. However, for the same genetic mutation, patients suffering from muscular dystrophy may exhibit significant differences in clinical signs. Symptoms may vary in nature, as well as in severity [21][22][23][24]. These observations led to the hypothesis that genetic modifiers may exist in muscular dystrophies, including ANXA.
In 2003, Bansal and collaborators revealed for the first time a direct link between a failure in membrane repair, caused by mutations in DYSF gene, and the development of a muscular dystrophy, e.g., LGMDR2 (2B) [8]. The fundamental role of ANXA in membrane repair questions their implication in the development of muscular dystrophies. In humans, no correlation has been made to date between muscular dystrophy and alteration of an ANXA gene. However, for the same genetic mutation, patients suffering from muscular dystrophy may exhibit significant differences in clinical signs. Symptoms may vary in nature, as well as in severity [79,126,127,128]. These observations led to the hypothesis that genetic modifiers may exist in muscular dystrophies, including ANXA.ANXA1 and ANXA2 interact with DYSF to mediate sarcolemma repair [25] and both ANXA have been reported to be upregulated in Italian [26], American [27], or Australian patients [28] suffering from dysferlinopathies. Overexpression of these ANXA is likely an attempt to counteract the absence of DYSF and restore cell membrane repair ability. It has been revealed that excess of ANXA2 that leaks from injured myofibers activates muscle-resident fibro/adipogenic precursors that differentiate into adipocytes, which gradually replace dysferlin-deficient myofibers leading to muscle degeneration [29]. ANXA2 may act, therefore, as a modifying factor which strongly influences, in a negative way, clinical consequences of dysferlinopathies. Overexpression of ANXA2 is also observed in DMD, Becker muscular dystrophy or LMGDR12 and shedding of ANX-positive vesicles have been shown in ANO5-knockout myofibers (LMGDR12), suggesting these diseases may result from fibrotic or adipogenic replacement of myofibers [30][26]. Recently, an increase of 32% in the expression of ANXA2 has been also observed in a rat model of desminopathy [31].
ANXA1 and ANXA2 interact with DYSF to mediate sarcolemma repair [40] and both ANXA have been reported to be upregulated in Italian [129], American [130], or Australian patients [131] suffering from dysferlinopathies. Overexpression of these ANXA is likely an attempt to counteract the absence of DYSF and restore cell membrane repair ability. It has been revealed that excess of ANXA2 that leaks from injured myofibers activates muscle-resident fibro/adipogenic precursors that differentiate into adipocytes, which gradually replace dysferlin-deficient myofibers leading to muscle degeneration [132]. ANXA2 may act, therefore, as a modifying factor which strongly influences, in a negative way, clinical consequences of dysferlinopathies. Overexpression of ANXA2 is also observed in DMD, Becker muscular dystrophy or LMGDR12 and shedding of ANX-positive vesicles have been shown in ANO5-knockout myofibers (LMGDR12), suggesting these diseases may result from fibrotic or adipogenic replacement of myofibers [54,129]. Recently, an increase of 32% in the expression of ANXA2 has been also observed in a rat model of desminopathy [133].ANXA1 and ANXA2 are susceptible to cleavage by calpains [32], which may be critical for their function in membrane repair [25][33][34]. In calpainopathies, such as LGMDR1 (2A) [10], therefore, it is expected that calpains deficiency may lead to misfunction of ANXA and impairment of membrane resealing.
ANXA1 and ANXA2 are susceptible to cleavage by calpains [134], which may be critical for their function in membrane repair [40,56,135]. In calpainopathies, such as LGMDR1 (2A) [10], therefore, it is expected that calpains deficiency may lead to misfunction of ANXA and impairment of membrane resealing.In addition, a loss of function of ANXA1 and ANXA6 is observed in LGMDR12 and DMD. In damaged ANO5-knockout myofibers (LGMDR12), accumulation of both ANXA is reduced, altering the tight repair cap structure [30]. In DMD, ANXA1 and ANXA6 present a reduced expression leading to exacerbated sarcolemmal injury and delayed repair cap formation due to overexpression of osteopontin [24].
In addition, a loss of function of ANXA1 and ANXA6 is observed in LGMDR12 and DMD. In damaged ANO5-knockout myofibers (LGMDR12), accumulation of both ANXA is reduced, altering the tight repair cap structure [54]. In DMD, ANXA1 and ANXA6 present a reduced expression leading to exacerbated sarcolemmal injury and delayed repair cap formation due to overexpression of osteopontin [128].The role played by ANXA6 as a genetic modifier of muscular dystrophies is definitely the most described. It has been reported that ANXA6 knockdown in a zebrafish model of dysferlinopathy reinforces the dystrophic phenotype [35]. In addition, a truncated form of ANXA6, named ANXA6N32, has been identified in Sgcg-null mouse, a model of LGMDR5 (2C) [36] and in dysferlinopathic mice [37]. ANXA6N32 dramatically impairs translocation of the full-length ANXA6 to the membrane disruption site, disrupts the protein scaffold that is pivotal for membrane resealing, and enhances muscular dystrophy [36][37].
The role played by ANXA6 as a genetic modifier of muscular dystrophies is definitely the most described. It has been reported that ANXA6 knockdown in a zebrafish model of dysferlinopathy reinforces the dystrophic phenotype [46]. In addition, a truncated form of ANXA6, named ANXA6N32, has been identified in Sgcg-null mouse, a model of LGMDR5 (2C) [136] and in dysferlinopathic mice [137]. ANXA6N32 dramatically impairs translocation of the full-length ANXA6 to the membrane disruption site, disrupts the protein scaffold that is pivotal for membrane resealing, and enhances muscular dystrophy [136,137].Finally, ANXA7 has been also reported as disturbed in skeletal muscle from patients suffering from DMD and MDX mouse, whereas normal muscle contains specifically a 51-kDa ANXA7 isoform, dystrophic muscle exhibits the additional 47-kDa isoform, usually found in undifferentiated myoblasts [38][39]. During progression of the disease, ANXA7 is gradually retrieved in higher concentration in the serum of patients, suggesting the absence of membrane resealing of injured myofibers and the leak of ANXA7 [38]. If its participation in sarcolemma repair remains to be established, ANXA7 has been shown to mediate membrane repair in cancer cells by enabling assembly of the ESCRT-III complex [40].
Finally, ANXA7 has been also reported as disturbed in skeletal muscle from patients suffering from DMD and MDX mouse, whereas normal muscle contains specifically a 51-kDa ANXA7 isoform, dystrophic muscle exhibits the additional 47-kDa isoform, usually found in undifferentiated myoblasts [49,118]. During progression of the disease, ANXA7 is gradually retrieved in higher concentration in the serum of patients, suggesting the absence of membrane resealing of injured myofibers and the leak of ANXA7 [49]. If its participation in sarcolemma repair remains to be established, ANXA7 has been shown to mediate membrane repair in cancer cells by enabling assembly of the ESCRT-III complex [48]. Understanding how ANXA can modify the evolution of muscular dystrophies remains a huge project. In particular, most hitherto carried-out studies have used animal models and some differences may exist in the etiology and severity of muscular dystrophies between humans and animals. It will be, therefore, interesting in the near future to be able to explore these questions in human skeletal muscle cells.