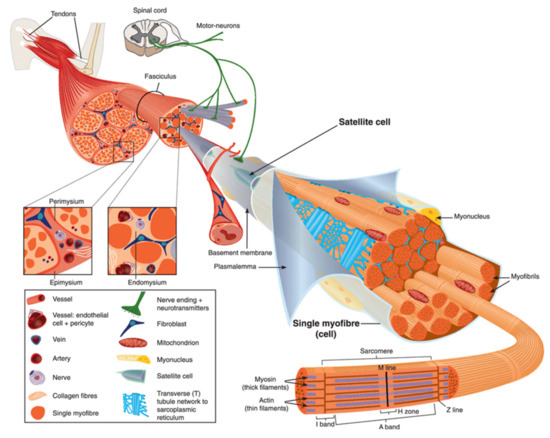
Human body is made up of approximately 650 muscles, with variability between individuals. Skeletal muscle tissue is itself made up of a collection of cell populations bounded by an envelope of fibrous connective tissue, called epimysium () [
15]. This envelope ensures the maintenance and protection of the muscle during contraction, and is also the link between muscle and bones. A muscle is subdivided into a set of muscle bundles which are made up of several dozen muscle cells, all of which are surrounded by a connective sheath, called perimysium (). Perimysium helps to structure the muscle and also to anchor muscle to bones at the level of tendons. The basal lamina or endomysium constitutes an extracellular matrix sheath that surrounds each muscle cell within a bundle (). This extracellular matrix, essentially composed of collagen, ensures the stability of the muscle fiber by interaction with the intracellular cytoskeleton. It also ensures the cohesion of muscle bundle by connecting neighboring muscle cells.
Figure 1. Scheme of skeletal muscle and associated structures. (
Left-hand part) Epimysium, perimysium, and endomysium constitute three connective tissue layers that form the lattice network and associated basement membranes in which myofibers regenerate after injury. The epimysium is the outer layer that surrounds the entire muscle and is contiguous with the tendon and endosteum (fascia surrounding bone). The perimysium surrounds bundles of myofibers. The endomysium is located between individual muscle fibers. (
Right-hand part) Satellite cells are located between basement membrane and sarcolemma. Sarcolemma bounds each myofiber, which is composed by multiple nuclei and the sarcoplasm that contains mitochondria, sarcoplasmic reticulum and myofibrils. The myofibril is the contractile unit of a myofiber. Specialized cytoskeleton within the myofibril forms repeated structures, called sarcomeres, which appear as a succession of light and dark bands under polarized light optical microscopy. The sarcoplasmic reticulum is the major provider of Ca
2+ required for muscle contraction. It is connected to transverse tubules that surround sarcomeres. Adapted from Reference [
15] with the Permission 5036470714502 from John Wiley and sons.
Skeletal muscle is made up of two main cell populations: skeletal muscle cells, also called muscle fibers or myofibers or myocytes, and mononuclear stem cells, called satellite cells (). Satellite cells are generally quiescent, with the ability to proliferate, differentiate and fuse to form new myofibers. These cells are considered to be the main contributor to post-natal muscle growth and maintenance [
16].
Myofibers are definitely special cells in terms of morphology and function. They exhibit a tubular and elongated shape and measure from a few hundred µm to several tens of centimeters in length for a diameter between 10 and 100 µm. Sarcolemma defines cytoplasm (sarcoplasm), which is composed by mitochondria, specialized endoplasmic (sarcoplasmic) reticulum, as well as a set of basic organelles, and a large number of nuclei located on the periphery of the cell.
Sarcolemma is subjected to severe mechanical stress, more than in any other cell type, due to its huge surface and to contraction and stretching processes that it regularly undergoes [
17]. To cope with these mechanical stresses, sarcolemma is supported by an exceptional protein framework, composed in particular of dystrophin, dystroglycans, and sarcoglycans [
18]. Unrepaired sarcolemma damage leads to the death of myofiber, which is followed by inflammation, especially through the infiltration of macrophages, and regeneration phases, during which the satellite cells proliferate, differentiate and fuse to form a new fiber [
19]. Satellite cells are not the only helping cells in skeletal muscle repair. Macrophages have been also reported to mediate sarcolemma repair through a mechanism involving dysferlin (DYSF) and phosphatidylserine (PS) [
20]. DYSF accumulated at the membrane disruption site, subsequently to influx of Ca
2+, promotes the accumulation of PS in the outer leaflet of the sarcolemma, which triggers the recruitment of macrophages. Excess of membrane and protein used during the resealing process and accumulated at the disruption site is engulfed by macrophages, which enables cell membrane integrity to be regained [
20].
3. ANXA and Muscular Dystrophies
In 2003, Bansal and collaborators revealed for the first time a direct link between a failure in membrane repair, caused by mutations in DYSF gene, and the development of a muscular dystrophy, e.g., LGMDR2 (2B) [
8]. The fundamental role of ANXA in membrane repair questions their implication in the development of muscular dystrophies. In humans, no correlation has been made to date between muscular dystrophy and alteration of an ANXA gene. However, for the same genetic mutation, patients suffering from muscular dystrophy may exhibit significant differences in clinical signs. Symptoms may vary in nature, as well as in severity [
79,
126,
127,
128]. These observations led to the hypothesis that genetic modifiers may exist in muscular dystrophies, including ANXA.
ANXA1 and ANXA2 interact with DYSF to mediate sarcolemma repair [
40] and both ANXA have been reported to be upregulated in Italian [
129], American [
130], or Australian patients [
131] suffering from dysferlinopathies. Overexpression of these ANXA is likely an attempt to counteract the absence of DYSF and restore cell membrane repair ability. It has been revealed that excess of ANXA2 that leaks from injured myofibers activates muscle-resident fibro/adipogenic precursors that differentiate into adipocytes, which gradually replace dysferlin-deficient myofibers leading to muscle degeneration [
132]. ANXA2 may act, therefore, as a modifying factor which strongly influences, in a negative way, clinical consequences of dysferlinopathies. Overexpression of ANXA2 is also observed in DMD, Becker muscular dystrophy or LMGDR12 and shedding of ANX-positive vesicles have been shown in ANO5-knockout myofibers (LMGDR12), suggesting these diseases may result from fibrotic or adipogenic replacement of myofibers [
54,
129]. Recently, an increase of 32% in the expression of ANXA2 has been also observed in a rat model of desminopathy [
133].
ANXA1 and ANXA2 are susceptible to cleavage by calpains [
134], which may be critical for their function in membrane repair [
40,
56,
135]. In calpainopathies, such as LGMDR1 (2A) [
10], therefore, it is expected that calpains deficiency may lead to misfunction of ANXA and impairment of membrane resealing.
In addition, a loss of function of ANXA1 and ANXA6 is observed in LGMDR12 and DMD. In damaged ANO5-knockout myofibers (LGMDR12), accumulation of both ANXA is reduced, altering the tight repair cap structure [
54]. In DMD, ANXA1 and ANXA6 present a reduced expression leading to exacerbated sarcolemmal injury and delayed repair cap formation due to overexpression of osteopontin [
128].
The role played by ANXA6 as a genetic modifier of muscular dystrophies is definitely the most described. It has been reported that ANXA6 knockdown in a zebrafish model of dysferlinopathy reinforces the dystrophic phenotype [
46]. In addition, a truncated form of ANXA6, named ANXA6N32, has been identified in Sgcg-null mouse, a model of LGMDR5 (2C) [
136] and in dysferlinopathic mice [
137]. ANXA6N32 dramatically impairs translocation of the full-length ANXA6 to the membrane disruption site, disrupts the protein scaffold that is pivotal for membrane resealing, and enhances muscular dystrophy [
136,
137].
Finally, ANXA7 has been also reported as disturbed in skeletal muscle from patients suffering from DMD and MDX mouse, whereas normal muscle contains specifically a 51-kDa ANXA7 isoform, dystrophic muscle exhibits the additional 47-kDa isoform, usually found in undifferentiated myoblasts [
49,
118]. During progression of the disease, ANXA7 is gradually retrieved in higher concentration in the serum of patients, suggesting the absence of membrane resealing of injured myofibers and the leak of ANXA7 [
49]. If its participation in sarcolemma repair remains to be established, ANXA7 has been shown to mediate membrane repair in cancer cells by enabling assembly of the ESCRT-III complex [
48].
Understanding how ANXA can modify the evolution of muscular dystrophies remains a huge project. In particular, most hitherto carried-out studies have used animal models and some differences may exist in the etiology and severity of muscular dystrophies between humans and animals. It will be, therefore, interesting in the near future to be able to explore these questions in human skeletal muscle cells.