Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Dong-Kug Choi | + 6181 word(s) | 6181 | 2021-05-14 05:44:14 | | | |
| 2 | Vivi Li | Meta information modification | 6181 | 2021-05-20 05:25:24 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Choi, D. G-Protein-Coupled Receptors in CNS. Encyclopedia. Available online: https://encyclopedia.pub/entry/9883 (accessed on 08 February 2026).
Choi D. G-Protein-Coupled Receptors in CNS. Encyclopedia. Available at: https://encyclopedia.pub/entry/9883. Accessed February 08, 2026.
Choi, Dong-Kug. "G-Protein-Coupled Receptors in CNS" Encyclopedia, https://encyclopedia.pub/entry/9883 (accessed February 08, 2026).
Choi, D. (2021, May 20). G-Protein-Coupled Receptors in CNS. In Encyclopedia. https://encyclopedia.pub/entry/9883
Choi, Dong-Kug. "G-Protein-Coupled Receptors in CNS." Encyclopedia. Web. 20 May, 2021.
Copy Citation
Neurodegenerative diseases are a large group of neurological disorders with diverse etiological and pathological phenomena. However, current therapeutics rely mostly on symptomatic relief while failing to target the underlying disease pathobiology. G-protein-coupled receptors (GPCRs) are one of the most frequently targeted receptors for developing novel therapeutics for central nervous system (CNS) disorders. Many currently available antipsychotic therapeutics also act as either antagonists or agonists of different GPCRs. Therefore, GPCR-based drug development is spreading widely to regulate neurodegeneration and associated cognitive deficits through the modulation of canonical and noncanonical signals.
serotonin
cannabinoid receptor
metabotropic glutamate receptor
seven transmembranes
orphan G-protein-coupled receptors
1. Introduction
G-protein-coupled receptors (GPCRs) constitute the largest family of membrane receptors in humans, and ~34% of marketed drugs target this family. Since their discovery, this receptor family has been both pharmacologically and biologically important in the treatment of different pathological conditions. GPCRs contain a common pattern in their basic structures. They consist of seven membrane-spanning domain structures, which they use to sense different signals and stimuli, including light, stress, hormones, peptides, and so forth [1].
Neurodegenerative diseases (NDDs) are most common and prevalent in elderly people worldwide [2] and cause progressive neuronal dysfunction, toxicities, and death. These diseases lead to an irreversible weakening of all brain functions. Around 30 million individuals worldwide are affected by NDDs [3][4]. Alzheimer’s disease (AD), vascular dementia (VaD), frontotemporal dementia (FTD), Parkinson’s disease (PD), and Huntington’s disease (HD) [5] are the prevalent NDDs commonly diagnosed in aged people. The cognitive function in patients is seriously affected during these disease’s progression. Therefore, the age-related decline in cognitive function is a leading challenge in mental health research [6]. Although some symptomatic treatments are available for NDDs, no specific treatments have yet been found [7]. Among the various NDDs, AD, which causes impaired cognitive function and memory loss, is the most extensively discussed and researched [8]. Amyloid β (Aβ) plaque formation and the hyperphosphorylation of tau proteins in the neurofibrillary tangles of the frontal and temporal lobe and other regions of the neocortex are observed during the progression of AD. VaD is another common cause of dementia, in which patients often complain of disturbances in frontal executive function [1] and multiple cerebrovascular pathologies, including vessel occlusion, arteriosclerosis, hypertension, aneurysms, and various forms of arteritis [8]. FTD can be diagnosed in people less than 65 years old [9] and is characterised by neuropsychiatric symptoms and behavioural, motor, and cognitive impairments [10]. Abnormal depositions of tau proteins are observed in different regions of an FTD patient’s brain, such as the hippocampus, frontal cortex, and striatum [11].
2. GPCRs’ Relevance in Cognition and Neurodegenerative Disorders
GPCRs comprise the largest family of transmembrane receptors, with over 800 members present in humans [12][13]. Slightly over half of the 800 receptors have sensory functions that mediate olfaction (400), taste (33), light perception (10), and pheromone signalling (5) [14]. More than 90% (370) of the remaining receptors are expressed in the brain. These nonsensory GPCRs mediate signalling from multiple ligand types and regulate several physiological processes throughout the human organism, mainly endocrine and neurological processes [8][12][15] (Figure 1). GPCRs are currently the most common therapeutic drug target [12]. In 2017, Hauser and coworkers accumulated data from the Center Watch Drugs in Clinical Trials database and, by cross-referencing with public sources, reported 475 approved drugs that target GPCRs, representing 34% of all FDA-approved drugs [12][16]. This report also indicates that about 20% to 50% of drugs have GPCRs as a target, with discrepancies probably resulting from the varying definitions of ‘drug target’ [15][17][18]. As new functions for GPCRs are discovered, especially in the case of the 100 orphan GPCRs for which no endogenous ligand or clearly defined function are currently known, the number of drugs targeting GPCRs is expected to increase [13][18]. Therefore, the relevance of the different types of GPCRs in neurodegeneration and age-related disease progression is the focus of this article.
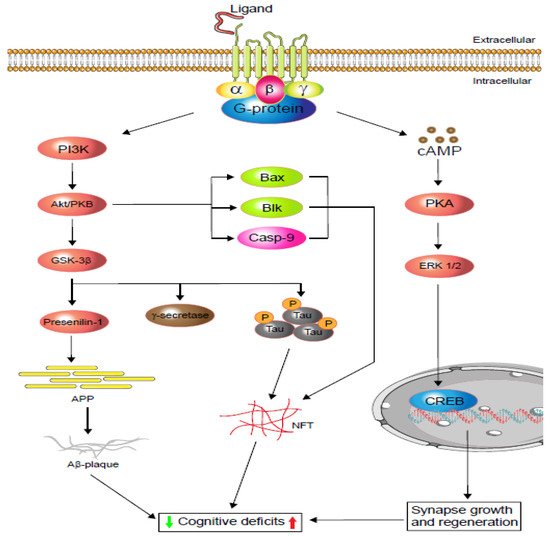
Figure 1. Schematic display of G-protein-coupled receptors (GPCRs) signalling in cognitive impairment. Depending on the agonist or inverse agonist ligand binding, the PI3/Akt-signalling pathway signals Bax and Casp-9 while increasing neurofibrillary tangle (NFT) formation via phosphorylation. PI3/Akt-signalling hyperphosphorylation also activates glycogen synthase kinase-3β (GSK-3β), which increases either or both tau protein phosphorylation and amyloid precursor protein (APP). Phosphorylated tau protein forms neurofibrillary tangles (NFTs) and regulates cognitive function. Similarly, APP metabolism regulates Aβ-plaque formation and controls cognition. Moreover, neuronal and dendritic plasticity is required for synaptic growth, regeneration, and memory formation, and it depends on extracellular signal-regulated kinase (ERK ½) modulation. Depending on the ligands, GPCRs activate cAMP-response element-binding protein (CREB) via the cAMP/ERK ½ pathway and regulate cognition (based on [19][20]).
2.1. Alzheimer’s Disease
AD is the predominant form of NDD and is most prevalent among people over the age of 65. AD patients exhibit cognitive deficits, memory loss, and changes in personality and behaviour [8][21]. The pathophysiology includes an accumulation of amyloid β (Aβ) plaques and the aggregation of tau proteins in neurofibrillary tangles, which are detected primarily in the frontal and temporal lobes and later slowly progress into other regions of the neocortex [21]. With the progression of this disease, different regions of the brain degenerate, and their neurochemical pathways change, including acetylcholine, serotonin and adenosine homeostasis, leading to cognitive impairments.
Although AD treatment strategies are progressing, they are limited to symptomatic relief only. One of the common features of AD is the gradual downregulation of the acetylcholine level in the brain, which is due to the abundance of acetylcholinesterase [22]. Acetylcholinesterase inhibitors (AChEi), therefore, play a vital role in the symptomatic treatment of AD and also improve cholinergic neurotransmission and cognitive function [23]. However, glutamatergic neurotransmitter overexpression is also reported in the pathogenesis of AD [24][25]. In addition, glutamate receptors are reported to mediate most of the excitatory neurotransmissions in the mammalian brain, and their overstimulation can cause excitotoxicity [24][26]. Metabotropic glutamate receptors (especially mGluR5) have been found to be involved in both cognition and Aβ generation and accumulation. A study showed that genetic deletion of mGluR5 in the mouse (APPswe/PS1ΔE9) ameliorates cognitive deficiencies and minimises Aβ production [27]. The study also indicates that mGluR5 expression is related to the increase in Aβ plaque and mediated by the mechanism of the fragile X mental retardation protein (FMRP) [27]. However, the mGluR5 antagonist 3-[(2-methyl-1,3-thiazol-4-yl) ethynyl]-pyridine (MTEP) has efficiently reversed this condition under the same paradigm [28]. Extensive evidence indicates that a wide-ranging serotonergic denervation of the neocortex and hippocampus occurs in AD patients. For example, activation of the serotonergic 5-HT2A and 5-HT4 receptors improves learning and memory [29][30] through the activation of the extracellular signal-regulated kinase (ERK) mediated by either the G-protein or β-arrestin (Figure 1) [19]. Therefore, targeting serotonergic receptors would also intervene in AD progression.
2.2. Vascular Dementia
Vascular dementia (VaD) is predominant in 8–15% of cognitively-impaired aged patients. Neuropathological investigation of VaD shows multifocal and/or diffuse lesions in the subcortical and strategically important brain areas, including the thalamus, frontobasal, and limbic system, and hippocampal sclerosis to multi-infarct encephalopathy [31]. These pathophysiologies further impair cognition, behaviour, execution, and memory. Further, mAChR1 reduction due to cerebrovascular occlusion results in cognitive dysfunction in VaD [32]. In addition, D1R reduction in the hippocampal dentate gyrus in a VaD rat model resulted in learning and memory deficits [33]. The γ-aminobutyric acid receptor (GABAR) in the dentate gyrus regulates synaptic plasticity, learning, and memory. In a VaD rat hippocampus, both GABAR1 and GABAR2 were found to be reduced, which resulted in spatial learning and memory deficits [34].
The 5-hydroxytryptamine receptors (5-HTRs) are prevalent in the frontal and temporal cortices [35] and are key players in cognitive function and memory formation. In particular, the 5-HT1A and 5-HT2A receptors are related to the correlation and preservation of cognition [36]. Reduced 5-HT1A receptors in AD brains leads to cognitive impairments [8][37] and could also affect VaD as well. Hence, although GPCRs have not yet been studied extensively in VaD models, GPCRs could be potential therapeutic targets in VaD as well.
2.3. Frontotemporal Dementia
Frontotemporal dementia (FTD) is a special type of dementia affecting both the front and sides of the brain. FTD causes verbal, executive, and behavioural impairments. FTD is mostly diagnosed in people younger than 65 [9], with the disease progression starting at ~45 years of age. As in other types of dementia, the patient’s condition worsens with age. FTD, in some cases, also has a microtubule-associated-protein-tau-based pathology. The mutations in the tau gene lead to different forms of tauopathy [38], including progressive supranuclear palsy and corticobasal degeneration, although most FTD patients do not show apparent tau gene or protein anomalies [38]. Clinical management of FTD is quite challenging, and it is important to understand the neurobiological substrate in order to ease the process. The role of GPCRs in FTD-induced cognitive deficits makes them an interesting target in terms of halting the disease’s progression.
FTD is a combination of impairments in neuropsychiatry and social behaviour. A recent study demonstrated that the neuropeptide oxytocin impacts social cognition and behaviour in FTD patients [39]. Oxytocin and vasopressin, another neuropeptide, are important in social cognition, trust, behaviour, and facial expressions. These neuropeptides are related to the pituitary and synthesised in the hypothalamus, primarily in large magnocellular neurons located in the supraoptic and paraventricular nuclei [40]. They are released into various brain regions, such as the amygdala and anterior cingulate cortex, for the paracrine signalling of the oxytocin receptor (OXTR) and vasopressin receptor (AVPR1a), and these receptors have a pathophysiological impact on FTD [41]. Intranasal vasopressin administration improves social cognition and facial expressions in humans, especially in the eye region [41]. Oxytocin administration in a similar paradigm also improved facial expressions and social interactions [41]. The presence of oxytocin also facilitates GABAergic synapse development, which blocks fear and anxiety [8][42].
Several serotonin receptors, including 5-HT1A and 5-HT2A, decline in the anterior cingulate cortex [43], orbitofrontal and medial prefrontal cortexes [44], and the frontal and temporal cortexes [43] of FTD patients. However, in AD patients, the levels of the 5-HT1A and 5-HT2A receptors fall in the hippocampus and prefrontal cortex region, which causes cognitive deficits. In contrast, under similar conditions, the FTD patient develops neuropsychiatric symptoms [37]. Moreover, the mGluR5 and NMDA receptors are colocalized in cortical regions of the brain and are also involved in the formation of receptor-dependent synaptic plasticity [45]. A recent study showed mGluR5 decreases in the paralimbic cortex region of FTD patients and may lead to the neurodegeneration observed [46]. Finally, mGluR5 activation enhances NMDA receptor action, while inhibition of the receptor blocks NMDA receptors [47].
2.4. Parkinson’s Disease
Parkinson’s disease (PD) is another prevalent neurodegenerative disease and is the second most ubiquitous age-linked disorder. The aetiology of PD includes motor and nonmotor dysfunctions [48][49][50]. One-quarter of PD patients are reported to develop mild cognitive impairment (MCI), with declines in memory (13.3%), visuospatial (11.0%), and attention/executive function (10.1%) [51]. A 20-year follow-up study of some newly diagnosed PD patients reported the occurrence of dementia in 83% of the cases [52]. However, most PD patients are diagnosed with elevated dopamine D1 and D2 receptors in the striatum and substantia nigra, which leads to dopamine denervation supersensitivity [53]. A study of PD animals showed that a D1 agonist improved visuospatial accuracy, whereas the use of a D2 antagonist induces attention impairment [54]. Therefore, the crucial roles of D1 and D2 in visuospatial and attentional dysfunction in PD–MCI are well supported. A good number of nondopaminergic transmitters are also affected in PD patients, and these transmitters are considered to contribute to cognitive deficits [55]. A post-mortem brain study of PD reported the loss of the cholinergic system, leading to cognitive decline. The cholinergic deficit is also associated with late-onset dementia in PD, whereas Aβ plaque and Lewy bodies are associated with an earlier onset of dementia [56][57]. Several serotonergic receptors are also found to be associated with cognition PD, such as the 5-HT1B receptor, which has been found to be downregulated in the early stage of PD progression [58].
2.5. Multiple Sclerosis
Within the context of multiple sclerosis’ (MS’s) pathophysiology, around 40–60% of patients complain of cognitive dysfunction [59]. Previously, it was supposed that depression and cognition are two independent domains that persist in MS. Eventually, it was understood that at a certain point in MS-induced depression, MS patients experience severe difficulties with working memory, execution, and information-processing time [60][61][62]. When proinflammatory cytokines in the central nervous system (CNS) increase, less serotonin is released from the synapses, causing the depression and resulting in the malfunction of the noradrenergic and serotonergic circuits. In this context, antidepressant therapeutics alone might not be sufficient to improve cognitive function in MS. For example, a study conducted in 2008 with two groups of MS patients reported only a slight difference between the group being treated with antidepressants and the placebo group. In that study, 57% of MS patients treated with paroxetine (a selective serotonin reuptake inhibitor (SSRI)) received 50% lower depression scores on the Hamilton Rating Scale, while the placebo group reduced their scores by 40% under similar conditions [63].
2.6. Huntington’s Disease
Progressive motor, behavioural, and cognitive impairment are the common features of Huntington’s disease (HD). Cognitive impairment is inevitable in HD. A great deal of evidence indicates that glutamate-mediated excitotoxicity, which includes wide mGluR2 and mGluR5 expression in the neocortical layers, hippocampus, striatum, thalamus/hypothalamus, and cerebellum, is one of the major contributors to HD progression [61][64][65]. It is also evident that mGluR regulates motor function and Huntington (HTT) protein aggregation in HD. A study showed that treatment with the mGluR5 antagonist 2-methyl-6-(phenylethynyl)-pyridine (MPEP) reduces hyperactivity and improves motor coordination in R6/2 HD transgenic mice [66]. Genetic deletion of mGluR5 has also been reported to improve motor function and reduce HTT protein aggregation in HdhQ111/Q111 knock-in mice [66]. On the other hand, both D1R and D2R expression declines in the early and late phase of HD [67]. The hyperactivity of dopamine in the early phase of HD, i.e., activation of D2R, increases HTT protein aggregation [68]. Thus, suppressing/inhibiting D2R with an antagonist would decrease HTT protein aggregation and reduce cell death via striatal protection, which may be beneficial in the treatment of early-stage HD and could potentially delay disease progression [69].
3. Therapeutic Targets for Neurodegenerative Disorders Based on Different GPCR Classes
Due to the selectivity in the function, specific distribution, and number of family members, GPCRs possess a potential therapeutic index and are important targets in different neurological conditions; some of the GPCRs listed in Table 1 are therapeutic targets for cognition and memory deficits. The important roles of different GPCRs are discussed below.
Table 1. Therapeutics for different neurodegenerative disorders—new agents, Phase II or III clinical trials [12].
| Agents | Targets | Receptor Family | Indications |
|---|---|---|---|
| Erenumab | CALCRL | Calcitonin | Migraine |
| Ubrogepant | CALCRL | Calcitonin | Migraine |
| Eptinezumab | CGRP | Calcitonin | Migraine |
| Cannabidivarin | GPR119 | GPR119 | MS and epilepsy |
| Cannabidiol | GPR55 | GPR55 | MS and epilepsy |
| VSN16R | GPR18 | GPR18 | MS and epilepsy |
| Δ-9-Tetrahydrocannabinol-cannabidiol (THC-CBD) | CB1/2 receptor | Cannabinoid | Spasticity in MS |
| Fingolimod | Sphingosine 1-phosphate (S1P) | Sphingosine 1-phosphate (S1P) receptor | MS |
| Xanomeline | M1/M4 | Cholinergic | AD |
| AF267B | M1 and M3 | mAChR | Reduces amyloid and tau pathologies in AD |
| Leuprolide | AChEI | Cholinergic | Synergizes AChEI activities |
| Vortioxetine | 5-HT3, 5-HT7, 5-HT1D | Serotonin | Major Depressive Disorder |
| Haloperidol | D2R | Dopamine | Working memory in PD |
| SK609 | D3R | Dopamine | Locomotor activity in PD |
| VCE-003.2 | CB2R | Cannabinoid | HD |
| DMXBA (GTS-21) and ABT-107 | α7nAChR | Cholinergic | PD |
| PHA 543613 | α7nAChR | Cholinergic | Early-stage HD |
| LY341495 | Group I/II mGluRs | mGluRs | Improves synaptic plasticity in AD |
| Galantamine | AChEI | Cholinergic | VaD, AD, PD, and Lewy bodies with dementia |
| Rivastigmine | AChEI | Cholinergic | VaD, AD, PD, and Lewy bodies with dementia |
3.1. 5-HT Receptors
The serotonergic neurotransmitters play a critical role in cognitive and behavioural functions. These receptors are distributed in different brain regions, including the cortex, amygdala, and hippocampus, and are associated with learning and memory [70]. Their roles in cognition and memory have made them important drug targets in neurodegenerative disorders. Some evidence suggests that the use of agonists of the 5-HT subtype, such as 5-HT2A/2C or 5-HT4, can prevent memory impairment and improve learning ability. A similar effect can also be obtained by using an antagonist of 5-HT1A or 5-HT3 and the 5-HT1B receptor [71].
The 5-HT6 receptor (5-HT6R) has been shown to regulate several neurotransmitter pathways, including the serotonergic, cholinergic, glutamatergic, and GABAergic systems [72]. In addition, several studies have established that this receptor is involved in learning and memory processes. These studies also claim that 5-HT6R antagonists could improve cognitive functions; some agents are in the preclinical stage [73][74]. Many of these agents are associated with substantial improvements in different cognitive tasks and enhanced memory retention or formation in rodents [75]. To date, at least three candidates have already reached Phase II/III clinical trials as novel therapeutic agents for the treatment of AD [76]. Indeed, the serotonin receptor 5-HT6R is an attractive drug target for reversing memory loss and learning disabilities associated with NDDs [76][77][78]. A recent study has discovered a new benzimidazole-based compound that is an antagonist of 5-HT6R and improves the novel object identification task in memory-deficit mice [75].
Moreover, in a Phase I trial, PRX-07034, a selective 5-HT6R antagonist, showed high selectivity for 5-HT6Rs over other 5-HT receptors and nonserotonin receptors. This derivative at doses of 1 and 3 mg/kg enhanced short-term memory and improved cognitive flexibility in rats [79]. Although Phase II trials have not yet been concluded [80][81], this drug candidate could be useful in treating dementia in AD [82]. Another selective antagonist, AVN-322, which is a derivative of AVN-101 and AVN-211, is undergoing Phase I trials for AD and schizophrenia. This drug has been reported to reverse the detrimental cognitive effects of scopolamine and MK-80 [83]. The AVN-101 is a multitarget serotonin antagonist that blocks 5-HT7R and has a low, but potential, affinity towards 5-HT6, 5-HT2A, 5-HT2C, and adrenergic receptors. It is also a dynamic candidate that is an antagonist of both 5-HT6 and 5-HT7. Both receptors follow the same signal transduction pathway, which is important for learning, memory, and anxiety. After completing Phase I trials, the drug is poised to start Phase II trials for AD and anxiety [84]. In addition, the benefit of AVN-101 in treating AD progression is the symptomatic relief of anxiety, depression, sleep disorders, and associated mood disorders [84]. Another derivative, AVN-211, has been reported to be a well-tolerated antagonist at a dose of 20 mg/kg of body weight and is suggested in AD therapy for its positive impact on cognition [85].
Idalopirdine is an antagonist of 5-HT6R that was developed initially for schizophrenia. Later analysis revealed its positive effect on cognition, and it was recommended for use in AD-associated dementia. Idalopirdine blocks 5-HT6R and increases acetylcholine in the CNS. In particular, it increases acetylcholine by inhibiting CYP206, an enzyme that is involved in the metabolism of donepezil. Therefore, cotreatment with donepezil increases donepezil’s bioavailability [86]. During a trial, 90 mg of idalopirdine taken daily with donepezil (10 mg) improved cognitive function significantly [86]. An AChE inhibitor, donepezil, and an NMDAR inhibitor, memantine, were used in a clinical trial involving VaD patients and resulted in cognitive improvement [87]. Monotherapy with intepirdine was found to be well tolerated, but its efficacy was lower than expected in AD. Further, the results of different studies were inconsistent. For instance, one study found a significant change in global function but not cognition [88], while another report failed to find any significant improvements [89][90].
Latrepirdine, or Dimebon, is an antihistamine developed originally for allergic rhinitis. Later, the drug was found to have procognitive effects, which are the result of antagonising 5-HT6R. Latrepirdine was also reported to stabilise mitochondria and works as a neuroprotective drug [81]. Based on fact and figures, latrepirdine was recommended for clinical trials in both AD and HD in the 2000s [81]. After completion of a small pilot study, the drug passed its Phase I and II clinical trials. However, in the Phase III trial, use of latrepirdine was terminated due to lack of efficacy [81][91]. SUVN-502 is another selective 5-HT6R antagonist. After completing Phase I trials with no notable adverse effects and better tolerance [92], the drug is currently in Phase II clinical trials involving patients with moderate AD [81]. This drug has been reported to promote acetylcholine and glutamate in the CNS when administered in combination with donepezil [93][94]. Agomelatine, a 5-HT2C antagonist, is another drug undergoing clinical trials that promises to improve prefrontal dopaminergic tone in FTD patients [95]. One clinical trial involving agomelatine reported that it reduces depression in AD patients significantly and decreases depression and motor symptoms in PD patients [96][97]. These data suggest that the 5-HT2C receptor could also be an important target in neurodegenerative disorder therapy.
Tryptophan (TRP) is an essential amino acid and serotonin precursor, and its depletion can disrupt serotonin synthesis in the brain. That depletion could also produce a paradigm of the serotonin deficit models, which has been demonstrated in different animals, including mice [98], rats [99], primates [100], and humans [101]. Several studies have assessed the relation between TRP depletion and cognitive function. For instance, Mendelsohn et al. [102] used a diet lacking in TRP but enriched in large neutral amino acids to produce an almost 50% downregulation of 5-HT levels in the cortex, striatum, and hippocampus [102]. That study, however, did not find any significant effect of TRP depletion on spatial, episodic, or working memory and instead reported semantic memory improvement even in the depleted state [102].
Moreover, despite the modest effect of TRP depletion on sustained attention, such as vigilance, it does not significantly affect specific impaired executive functions such as planning, decision making, and responding [102][103]. To the contrary, tryptophan-rich supplementation of a regular diet can enhance serotonin synthesis, and a clinical study found that tryptophan supplementation improved reaction times, visual memory, and attention [104]. Chronic ingestion of docosahexaenoic acid phospholipids with melatonin and tryptophan for 12 weeks also improves mild cognition impairment in elderly patients [105].
Vortioxetine [12], a serotonin transporter inhibitor (SERT), also works as an antagonist of 5-HT3, 5-HT7, and 5-HT1D receptors, an agonist of 5-HT1A, and a partial agonist of 5-HT1B. A recent meta-analysis of a 6–8-week treatment with vortioxetine (5-20 mg/day) produced an incremental reduction in depression symptoms, and an increasing effect was associated with an increase in dose [106]. Vortioxetine was also shown to improve cognitive performance in patients with acute major depressive disorder [106]. Similarly, a study involving chronic citalopram use (20 mg; single dose daily) resulted in improved impulse response and contextual information processing abilities on a delayed nonmatching to sample task (DNMST) in healthy controls (n = 20). Acute administration (24 hr) of that drug showed no effect on working memory or impulsive responses. The authors of the study also suggested that DNMST makes a contribution to the activation of 5-HT1A receptors in the entorhinal cortex and hippocampus [107].
Chronic consumption of a diet high in saturated fats and low in fibre is associated with obesity and cognitive decline. In this context, dietary supplementation can prevent cognitive decline by altering serotoninergic signalling in the brain. A recent study with high-fat, low-fibre (5% dietary fibre)-induced obese models has demonstrated an association with the upregulation of 5-HT1AR and 5-HT2AR binding density in the rat brain in comparison to the low-fat diet group [108]. With the inclusion of galacto-oligosaccharides and resistant starch, receptor binding densities in the hippocampal and hypothalamic region are reduced, improving cognitive function.
3.2. Dopamine Receptors
In the late 1950s, Carlsson identified dopamine as a potential neurotransmitter in the brain. Later, it was discovered that a progressive decrease in dopamine is associated with the pathophysiology of PD [50][109][110]. This finding introduced levodopa, the metabolic precursor of dopamine, into the symptomatic treatment of PD. In the past few years, several investigations have been conducted on dopamine and cognitive function [111][112][113][114]. Although dopamine neurons are very few in number compared to the total neuronal population in the brain (< 1/100,000), they are involved in neuroendocrine regulation, mood, motivation, and psychological processes, including working memory and learning [110][111]. Although no specific mechanism has yet been confirmed, the inhibition of tyrosine hydroxylase activity and tyrosine conversion into dopamine and norepinephrine are involved in long-term memory formation [115]. For example, vasopressin is a hypothalamic neuropeptide found to improve memory and does so by interacting with dopamine in the amygdala and serotonin of the hippocampus [116].
As previously mentioned, some evidence suggests that dopamine modulates working memory, but its specific role is not yet fully defined, and its effect in memory processing has not been clarified. Haloperidol, a D2R antagonist, has recently been investigated for its effect on working memory improvement as well as for its distracting-information-ignoring capability. The study was designed with two testing sessions. In one session, participants took a placebo tablet, and in the other, they took Haloperidol 2.5 mg [117]. The study showed that the deleterious effect of haloperidol on response conflict is associated with the negative effect of the drug on ignoring. The authors also suggest that D2R protects memory content from distraction through a general process, and inhibition of D2R could result in the impairment of response conflict as well as reduced quality of recall [117]. D1R and D2R have been found to be important targets in FTD as well. Both DR antagonists (antipsychotics) and agonists (specifically, D2R) are used frequently in FTD treatment. Commonly used antipsychotics, such as olanzapine, quetiapine, and risperidone, have a high affinity for D2R and dissociate rapidly, resulting in very few side effects [118]. A lack of presynaptic dopaminergic nerve terminal and postsynaptic D2R binding in the striatum is prevalent in FTD patients, and most of them complain of rigidity and bradykinesia. Therefore, FTD patients are currently being treated with the DR agonists carbidopa and levodopa to improve behavioural and psychotic symptoms.
However, SK609, a recently designed selective-small-molecule agonist of D3R, selectively inhibits norepinephrine reuptake as well as increases dopamine by 160% at an i.p. (intraperitoneal) dose of 4 mg/kg. SK609 improved rats’ performance in an attention task. Additionally, SK609 has been reported to improve cognitive function in low-performing rats. Interestingly, the molecule did not produce any side effects mediated by DA transporter (DAT) activity [119], such as spontaneous locomotor activity.
3.3. Cannabinoid Receptors
Extensive evidence indicates that the endocannabinoid system has modulatory effects on cognitive function, and these effects have a substantial role in memory acquisition, consolidation, and extinction [120]. However, the introduction of cannabinoid drugs often induces opposite effects, when used in anxiety, cognition, and other behavioural deficiencies, depending on the stress level and the aversiveness of the context [120][121]. It has been demonstrated that the use of an endocannabinoid receptor inhibitor under different environmental conditions has a substantial influence on cognitive function without affecting locomotor or emotional behaviour [121]. Although it is difficult to define the exact role of the cannabinoid receptor, cannabinoid signalling influences memory processing. This influence has been demonstrated in several studies [120][121][122].
In particular, Δ9-tetrahydrocannabinol (THC) is a cannabinoid receptor agonist that acts at CB1R to produce a wide range of biological and behavioural activity. This ligand’s association with cognitive deficits and poor decision making has been identified. Using Wistar rats in a rat gambling task study, it has been reported that a high dose of THC reduces premature responses, while another synthetic agonist produced the opposite reaction [123]. Although acute or limited chronic use of THC does not affect subjects, long-term exposure can impair impulse control and attentional function [124]. Therefore, chronic activation of CB1R or antagonism can impair or improve task performance, which makes it an interesting target. For example, in an animal study, the CB1 antagonist rimonabant produced an improvement in decision making [123]. Meanwhile, cannabinoid receptor agonists, such as AEA (N-arachidonoyl ethanolamine) and noladin, have been reported to protect against Aβ-induced neurotoxicity in the human teratocarcinoma cell line NTERA-2/cl-D1. The effect was exerted through the CB1R- and mitogen-activated protein kinase (MAPK) pathway [125]. Treatment with the CB1R antagonist SR141716A failed to protect against Aβ-induced amnesia [126]. Another study with a triple transgenic mouse model of AD (3xTg-AD) reported that CB1R is up-regulated in the anterior thalamus at the age of 4 months, while the CB1R activity decreased gradually in the nucleus basalis of Meynert at 15 months of age [127].
Activation of CB1R and CB2R was reported to affect the upregulation of PPARγ signalling in an animal model study [128], where Aβ-induced neuroinflammation, neurodegeneration, and spatial memory impairment was attenuated. Activation of CB2R using a lower-dose agonist, JWH-015, eradicated native Aβ from human tissue and cleared a synthetic pathogenic Aβ peptide in a human macrophage cell line (THP-1). This effect was attributed to the antagonist SR144528, which is selective for CB2R, and the plaque removal effect induced by JWH-015 was reversed [129]. A selective CB2R agonist 1-((3-benzyl-3-methyl-2,3-dihydro-1-benzofuran-6-yl) carbonyl) piperidine (MDA7) has shown a modulatory effect on cognitive impairment induced by bilateral microinjection of Aβ (1-40) fibrils into the hippocampal CA1 area of rats. Intraperitoneal treatment of MDA7 (15 mg/kg) for 14 days reduced microglial CD11b expression, promoted Aβ clearance, and restored synaptic plasticity, cognition, and memory [130].
The increased expression of CB2R has been noted with reduced CB1R in late-onset HD, mainly in glial cells [131]. The selective CB2R antagonist, SR144528, provides striatal neuron protection in HD rats; the mechanism includes glial cells [132]. Genetic deletion of CB2R also exacerbates HD, but CB2R-selective agonists can reduce striatal neurodegeneration through microglial activation [133]. SR141716 has also been reported to aggravate malonate-induced striatal pathology in HD rats [134]. However, some studies report that cannabidiol, a phytoconstituent and an allosteric modulator of CB1R [135], can rescue neuronal loss induced by THC by preventing THC-induced CB1R loss [136]. Similarly, VCE-003.2, a cannabigerol derivative, improved the antioxidant barrier in 3-nitropropionic-acid-induced HD mice brains [137]. This agent also showed neuroprotection in SOD1G93A transgenic amyotrophic lateral sclerosis (ALS) mice by targeting CB2R and inhibiting endocannabinoid inactivation [138]. Based on different reports, it is speculated that CBR antagonists may provide a better therapeutic insight into HD treatment.
3.4. Cholinergic Receptors
Cholinergic receptors are divided into two classes, including the muscarinic and nicotinic receptor families. These two families are further subclassed according to the occurrence of many ACh receptor subtypes, and their differential dendritic, somatic, axonal, and synaptic localisation contributes to the varied roles that these receptors play in the CNS [139]. Five subtypes of mAChRs (M1 to M5) have been defined and pharmacologically characterized in the CNS. Their expression has been detected at very high levels in the subcortical structures and the cerebral cortex [140]. A modest level of mAChR expression was also reported in the frontal cortex, parietal cortex, temporal cortex, entorhinal cortex, occipital cortex, and insular and cingulate cortexes, with the highest values recorded for the temporal and occipital cortexes [140]. M1 receptors are the most abundantly expressed mAChRs [140].
Nicotinic AChRs (nAChRs) constitute the second cholinergic receptor type. This type of receptor relies on ligand-gated ion channels and contains five subunits, which are assembled into homomeric or heteromeric subunit combinations. The pharmacological and biological characteristics of these receptors are determined by this combination [141]. The nAChR subunits are composed of α4, α6, α7, β2, and β3 subunits and are mostly expressed in the striatum [142][143]. They are expressed on glutamatergic and dopaminergic neuron terminals, GABAergic interneurons, and cholinergic interneurons (ChIs) but are not present in medium spiny neurons (MSNs) [144][145]. However, nAChRs can express in both pre- and post-synaptic neurons. Thus, they can depolarise and increase excitability, causing glutamate, DA, and GABA to release [146][147].
The nAChRs form good targets in the treatment of neurodegeneration. In recent years, several agonists and partial agonists of the α7 subunit have been evaluated in the treatment of NDDs. Daily nicotine administration in a PD animal model produced improvements in motor coordination. That treatment also showed a beneficial effect on neuronal survival, as well as on microglial and astrocytic activation, providing neuroprotection against MPTP/MPP+ toxicity [148]. DMXBA (GTS-21) and ABT-107, both α7nAChR agonists, have also produced beneficial effects by attenuating nigrostriatal damage in 6-hydroxydopamine (6-OHDA)-induced rats [50]. Another agonist, PHA 543613, attenuates early-stage HD induced by striatal quinolinic acid lesions. PHA 543613 reduced microglial activation to protect neurons [149]. In another study, α7nAChR-agonist-treated 3xTg-AD mice showed improved cognitive function [150]. However, PNU-282987 has also produced improved motor activity, anxiety, and learning and memory development in the B6C3-Tg mice model for AD [151]. Similarly, two other agonists, AR-R17779 and ABBF (N-[(3R)-1-azabicyclo [2.2.2]oct-3-yl]-7-[2-(methoxy) phenyl]-1-benzofuran-2-carboxamide), are also reported to improve learning and memory [152][153].
ANAVEX2-73, a σ1R (sigma 1 receptor) agonist and muscarinic receptor ligand, has been studied on the Aβ25-35-injected AD mice model [154]. Because Aβ25-35 injection modulates mitochondrial respiration in the hippocampus, ANAVEX2-73 (0.01-1 mg/kg IP) restored respiration to normal and prevented Aβ25-35-induced increases in lipid peroxidation levels, Bax/Bcl-2 ratio, and cytochrome c release into the cytosol [154].
3.5. Metabotropic Glutamate Receptors
Metabotropic glutamate receptors (mGluRs) are members of the C GPCR family, and they consist of eight subtypes (mGlu1 to mGlu8) that are further subdivided into three groups depending on their amino acid sequences, G-protein coupling, and pharmacological characteristics. Group I consists of mGlu1 and mGlu5 receptors, which are coupled to Gq/G11 [155]. The Group II (mGlu2, mGlu3) and Group III (mGlu4, mGlu6, mGlu7, mGlu8) subtypes are coupled to Gi/Go. Both of these groups regulate adenylate cyclase negatively and can also activate the MAP kinase and PI-3-kinase pathways [155][156]. However, overexpression of mGlu5 has been reported in different neurodegenerative disorders [156]. Specifically, Aβ plaques have been found in the surroundings of astrocytes, spinal cord lesions, and MS lesions, ALS, PD, and in the hippocampal astrocytes of Down syndrome patients [157][158].
Several mGluRs agonists, antagonists, and positive and negative allosteric modulators have been studied using different neurodegenerative animal models. Collectively, these data on mGluRs provide an insight into the development of therapeutics for treating NDDs. However, LY341495 (an antagonist to Group I/II mGluR) was reported as blocking Aβ-enhanced long-term depression and improving synaptic plasticity [159]. The same study also reported that pretreatment with an mGluR1/5 agonist, 3,5-dihydroxyphenylglycine (DHPG), decreased Aβ-enhanced long-term depression [159]. SIB1757, a noncompetitive antagonist of mGluR5, prevented Aβ oligomer-induced synaptic N-Methyl-D-aspartic acid receptor NMDAR reduction [160]. A comparison study targeting Group II mGluRs showed that an mGlu2R positive allosteric modulator, N-4′-cyano-biphenyl-3-yl)-N-(3-pyridinylmethyl)-ethanesulfonamide hydrochloride (LY566332), amplified Aβ-induced neurodegeneration [161]. Treatment with the antagonist (2S,1′S,2′S)-2-(9-xanthylmethyl)-2-(2′-carboxycyclopropyl) glycine (LY341495) of mGlu2/3R prevented this effect. Similarly, the dual mGlu2/3 receptor agonist (−)-2-oxa-4-aminobicyclo [3.1.0] exhane-4,6-dicarboxylic acid (LY379268) exhibited neuroprotection via a paracrine mechanism mediated by transforming growth factor-β1 [161]. Therefore, dual activation of mGlu2R and mGlu3R may be a prime target for providing neuroprotection against Aβ-induced toxicity, and negative modulation of mGlu5R would also be a good target for PD and AD treatment.
3.6. Orphan GPCRs
More than 140 GPCRs remain mysterious and are referred to as ‘orphans’ (oGPCRs); most of them do not have any known ligands. Although very little is known about their endogenous or exogenous ligands, these so-called oGPCRS have gained considerable attention as drug targets [162]. Several findings have postulated the critical role of oGPCRs in the cognitive deficits in disorders such as AD and schizophrenia [163][164][165][166]. For example, an expression map of the mouse brain has listed 78 oGPCRs and showed that many of them are relevant to cognition, motivation, and emotional processing [167]. That study reported that oGPCRs, such as GPR17, GPR27, GPR37, GPR39, GPR63, GPR85, GPR88, GPR123 (Adgra1), GPR125 (Adgra3), GPR153, GPR176, and GPRc5c, are highly expressed in the prefrontal cortex (PFC) region of the mouse brain, which is involved in cognition and learning. Both mouse and human brain analyses have shown that four oGPCRs are highly expressed, including GPR88, GPR123, GPR149, and GPR151, but information about these oGPCRs is minimal, with the exception of GPR88 [167].
However, GPR3 has exhibited stable and enhanced expression throughout the ageing process in ten different regions of the healthy human brain, which has been linked to AD pathogenesis in multiple cohort studies [165]. Additionally, GPR3, independent of G-protein coupling, recruits β-arrestin 2 and promotes γ-secretase activity, which increases Aβ precursor protein (APP) cleavage and accelerates Aβ peptide production and accumulation [168]. Thus, deletion of GPR3 in the AD mouse alleviated cognitive impairment and restored memory [165]. Although GPR158 improves memory through transducing osteocalcin and regulating IP3 and BDNF in the CA3 neurons [166], GPR158 overexpression in PFC has a potential role in depressive behaviour, which is reversed by its depletion [169].
GPR52 is a nonodorant GPCR and is colocalized with both D1 and D2 receptors in the basal ganglia neurons. Histological experiments suggest that GPR52 promotes and regulates the Cre-lox system and may also modulate cognition and emotion, as it has involvement with both dopaminergic and glutaminergic neurotransmission [170]. Thus, the incorporation of the GPR52 antagonist may potentiate cognitive improvement and exert anxiolytic activity in psychiatric disorders [170]. GPR3 knockout also produced anxiety and depressive behaviour, with no noticeable locomotor impairment under stressful conditions. However, the lack of GPR3 has no preventive action in the learning involved in fear memory in a similar stressful condition in mice [171]. GPR3 also regulates serotonin (5-HT) and dopamine (DA) synthesis and reuptake, which makes it a primary target as well. A study has reported the possibility that serotonin reduction in the frontal cortex and hippocampus causes aggressive behaviours in GPR3 knockout mice [171]. This finding indicates that GPR3 modulates the serotonergic and dopaminergic system, which makes it a potential target in the therapy of AD or schizophrenia.
GPR55 is highly expressed in the pyramidal cells in the hippocampal CA1 and CA3 layers and modulates the synaptic plasticity of pyramidal cells [172]. However, GPR85 is highly expressed in the dentate gyrus region of the hippocampus [173][174] and prominently expresses in the phases of neuronal differentiation in the developing cerebral cortex [175]. This expression suggests a possible role of GPR85 in cognition, and this receptor could become a potential drug target as well.
References
- Sachdev, P.; Kalaria, R.; O’Brien, J.; Skoog, I.; Alladi, S.; Black, S.E.; Blacker, D.; Blazer, D.G.; Chen, C.; Chui, H.; et al. Diagnostic criteria for vascular cognitive disorders: A VASCOG statement. Alzheimer Dis. Assoc. Disord. 2014, 28, 206–218.
- Przedborski, S.; Vila, M.; Jackson-Lewis, V. Neurodegeneration: What is it and where are we? J. Clin. Investig. 2003, 111, 3–10.
- Jakaria, M.; Azam, S.; Cho, D.-Y.; Haque, M.E.; Kim, I.-S.; Choi, D.-K. The Methanol Extract of Allium cepa L. Protects Inflammatory Markers in LPS-Induced BV-2 Microglial Cells and Upregulates the Antiapoptotic Gene and Antioxidant Enzymes in N27-A Cells. Antioxidants 2019, 8, 348.
- Jakaria, M.; Azam, S.; Jo, S.-H.; Kim, I.-S.; Dash, R.; Choi, D.-K. Potential Therapeutic Targets of Quercetin and Its Derivatives: Its Role in the Therapy of Cognitive Impairment. J. Clin. Med. 2019, 8, 1789.
- Jakaria, M.; Kim, J.; Karthivashan, G.; Park, S.-Y.; Ganesan, P.; Choi, D.-K. Emerging signals modulating potential of ginseng and its active compounds focusing on neurodegenerative diseases. J. Ginseng Res. 2019, 43, 163–171.
- Jakaria, M.; Haque, M.E.; Kim, J.; Cho, D.Y.; Kim, I.S.; Choi, D.K. Active ginseng components in cognitive impairment: Therapeutic potential and prospects for delivery and clinical study. Oncotarget 2018, 9, 33601–33620.
- Jakaria, M.; Azam, S.; Haque, M.E.; Jo, S.-H.; Uddin, M.S.; Kim, I.-S.; Choi, D.-K. Taurine and its analogs in neurological disorders: Focus on therapeutic potential and molecular mechanisms. Redox Biol. 2019, 24, 101223.
- Huang, Y.; Todd, N.; Thathiah, A. The role of GPCRs in neurodegenerative diseases: Avenues for therapeutic intervention. Curr. Opin. Pharmacol. 2017, 32, 96–110.
- Warren, J.D.; Rohrer, J.D.; Rossor, M.N. Clinical review. Frontotemporal dementia. BMJ Clin. Res. Ed. 2013, 347, f4827.
- Jicha, G.A.; Nelson, P.T. Management of frontotemporal dementia: Targeting symptom management in such a heterogeneous disease requires a wide range of therapeutic options. Neurodegener. Dis. Manag. 2011, 1, 141–156.
- Gabryelewicz, T.; Masellis, M.; Berdynski, M.; Bilbao, J.M.; Rogaeva, E.; St George-Hyslop, P.; Barczak, A.; Czyzewski, K.; Barcikowska, M.; Wszolek, Z.; et al. Intra-familial clinical heterogeneity due to FTLD-U with TDP-43 proteinopathy caused by a novel deletion in progranulin gene (PGRN). J. Alzheimers Dis. JAD 2010, 22, 11–1133.
- Hauser, A.S.; Attwood, M.M.; Rask-Andersen, M.; Schiöth, H.B.; Gloriam, D.E. Trends in GPCR drug discovery: New agents, targets and indications. Nat. Rev. Drug Discov. 2017, 16, 829–842.
- De Oliveira, P.G.; Ramos, M.L.S.; Amaro, A.J.; Dias, R.A.; Vieira, S.I. Gi/o-Protein Coupled Receptors in the Aging Brain. Front. Aging Neurosci. 2019, 11.
- Mombaerts, P.J. Genes and ligands for odorant, vomeronasal and taste receptors. Nat. Rev. Neurosci. 2004, 5, 263–278.
- Heng, B.C.; Aubel, D.; Fussenegger, M. An overview of the diverse roles of G-protein coupled receptors (GPCRs) in the pathophysiology of various human diseases. Biotechnol. Adv. 2013, 31, 1676–1694.
- Sriram, K.; Insel, P.A.J. G protein-coupled receptors as targets for approved drugs: How many targets and how many drugs? Mol. Pharmacol. 2018, 93, 251–258.
- Rask-Andersen, M.; Masuram, S.; Schiöth, H.B. The druggable genome: Evaluation of drug targets in clinical trials suggests major shifts in molecular class and indication. Annu. Rev. Pharmacol. Toxicol. 2014, 54, 9–26.
- Laschet, C.; Dupuis, N.; Hanson, J.J. The G protein-coupled receptors deorphanization landscape. Biochem. Pharmacol. 2018, 153, 62–74.
- Barthet, G.; Framery, B.; Gaven, F.; Pellissier, L.; Reiter, E.; Claeysen, S.; Bockaert, J.; Dumuis, A. 5-hydroxytryptamine 4 receptor activation of the extracellular signal-regulated kinase pathway depends on Src activation but not on G protein or beta-arrestin signaling. Mol. Biol. Cell 2007, 18, 1979–1991.
- Maletínská, L.; Popelová, A.; Železná, B.; Bencze, M.; Kuneš, J. The impact of anorexigenic peptides in experimental models of Alzheimer’s disease pathology. J. Endocrinol. 2019, 240, R47.
- Serrano-Pozo, A.; Frosch, M.P.; Masliah, E.; Hyman, B.T. Neuropathological alterations in Alzheimer disease. Cold Spring Harb. Perspect. Med. 2011, 1, a006189.
- Colović, M.B.; Krstić, D.Z.; Lazarević-Pašti, T.D.; Bondžić, A.M.; Vasić, V.M. Acetylcholinesterase inhibitors: Pharmacology and toxicology. Curr. Neuropharmacol. 2013, 11, 315–335.
- Selkoe, D.J. The therapeutics of Alzheimer’s disease: Where we stand and where we are heading. Ann. Neurol. 2013, 74, 328–336.
- Dong, X.X.; Wang, Y.; Qin, Z.H. Molecular mechanisms of excitotoxicity and their relevance to pathogenesis of neurodegenerative diseases. Acta Pharmacol. Sin. 2009, 30, 379–387.
- Farlow, M.R.; Graham, S.M.; Alva, G. Memantine for the treatment of Alzheimer’s disease: Tolerability and safety data from clinical trials. Drug Saf. 2008, 31, 577–585.
- Willard, S.S.; Koochekpour, S. Glutamate, glutamate receptors, and downstream signaling pathways. Int. J. Biol. Sci. 2013, 9, 948–959.
- Hamilton, A.; Esseltine, J.L.; DeVries, R.A.; Cregan, S.P.; Ferguson, S.S. Metabotropic glutamate receptor 5 knockout reduces cognitive impairment and pathogenesis in a mouse model of Alzheimer’s disease. Mol. Brain 2014, 7, 40.
- Um, J.W.; Kaufman, A.C.; Kostylev, M.; Heiss, J.K.; Stagi, M.; Takahashi, H.; Kerrisk, M.E.; Vortmeyer, A.; Wisniewski, T.; Koleske, A.J.; et al. Metabotropic glutamate receptor 5 is a coreceptor for Alzheimer abeta oligomer bound to cellular prion protein. Neuron 2013, 79, 887–902.
- Lo, A.C.; De Maeyer, J.H.; Vermaercke, B.; Callaerts-Vegh, Z.; Schuurkes, J.A.; D’Hooge, R. SSP-002392, a new 5-HT4 receptor agonist, dose-dependently reverses scopolamine-induced learning and memory impairments in C57Bl/6 mice. Neuropharmacology 2014, 85, 178–189.
- Zhang, G.; Ásgeirsdóttir, H.N.; Cohen, S.J.; Munchow, A.H.; Barrera, M.P.; Stackman, R.W., Jr. Stimulation of serotonin 2A receptors facilitates consolidation and extinction of fear memory in C57BL/6J mice. Neuropharmacology 2013, 64, 403–413.
- Jellinger, K.A. The pathology of “vascular dementia”: A critical update. J. Alzheimers Dis. JAD 2008, 14, 107–123.
- Tanaka, K.-I.; Ogawa, N.; Asanuma, M.; Kondo, Y.; Nomura, M.J. Relationship between cholinergic dysfunction and discrimination learning disabilities in Wistar rats following chronic cerebral hypoperfusion. Brain Res. 1996, 729, 55–65.
- Wan, P.; Wang, S.; Zhang, Y.; Lv, J.; Jin, Q.J. Involvement of dopamine D1 receptors of the hippocampal dentate gyrus in spatial learning and memory deficits in a rat model of vascular dementia. Die Pharm. Int. J. Pharm. Sci. 2014, 69, 709–710.
- Li, C.-J.; Lu, Y.; Zhou, M.; Zong, X.-G.; Li, C.; Xu, X.-L.; Guo, L.-J.; Lu, Q.J. Activation of GABA B receptors ameliorates cognitive impairment via restoring the balance of HCN1/HCN2 surface expression in the hippocampal CA1 area in rats with chronic cerebral hypoperfusion. Mol. Neurobiol. 2014, 50, 704–720.
- Glikmann-Johnston, Y.; Saling, M.M.; Reutens, D.C.; Stout, J.C.J. Hippocampal 5-HT1A receptor and spatial learning and memory. Front. Pharmacol. 2015, 6, 289.
- Elliott, M.S.; Ballard, C.G.; Kalaria, R.N.; Perry, R.; Hortobagyi, T.; Francis, P.T. Increased binding to 5-HT1A and 5-HT2A receptors is associated with large vessel infarction and relative preservation of cognition. Brain 2009, 132, 1858–1865.
- King, M.V.; Marsden, C.A.; Fone, K.C.J. A role for the 5-HT1A, 5-HT4 and 5-HT6 receptors in learning and memory. Trends Pharmacol. Sci. 2008, 29, 482–492.
- Neary, D.; Snowden, J.; Mann, D. Frontotemporal dementia. Lancet Neurol. 2005, 4, 771–780.
- Jesso, S.; Morlog, D.; Ross, S.; Pell, M.D.; Pasternak, S.H.; Mitchell, D.G.; Kertesz, A.; Finger, E.C. The effects of oxytocin on social cognition and behaviour in frontotemporal dementia. Brain J. Neurol. 2011, 134, 2493–2501.
- Baribeau, D.A.; Anagnostou, E. Oxytocin and vasopressin: Linking pituitary neuropeptides and their receptors to social neurocircuits. Front. Neurosci. 2015, 9, 335.
- Donaldson, Z.R.; Young, L.J. Oxytocin, Vasopressin, and the Neurogenetics of Sociality. Science 2008, 322, 900–904.
- Theodosis, D.T.; Koksma, J.-J.; Trailin, A.; Langle, S.L.; Piet, R.; Lodder, J.C.; Timmerman, J.; Mansvelder, H.; Poulain, D.A.; Oliet, S.H.R.; et al. Oxytocin and estrogen promote rapid formation of functional GABA synapses in the adult supraoptic nucleus. Mol. Cell. Neurosci. 2006, 31, 785–794.
- Franceschi, M.; Anchisi, D.; Pelati, O.; Zuffi, M.; Matarrese, M.; Moresco, R.M.; Fazio, F.; Perani, D. Glucose metabolism and serotonin receptors in the frontotemporal lobe degeneration. Ann. Neurol. 2005, 57, 216–225.
- Bowen, D.; Procter, A.; Mann, D.; Snowden, J.; Esiri, M.; Neary, D.; Francis, P. Imbalance of a serotonergic system in frontotemporal dementia: Implication for pharmacotherapy. Psychopharmacology 2008, 196, 603–610.
- Alagarsamy, S.; Rouse, S.T.; Junge, C.; Hubert, G.; Gutman, D.; Smith, Y.; Conn, P.J. NMDA-induced phosphorylation and regulation of mGluR5. Pharmacol. Biochem. Behav. 2002, 73, 299–306.
- Leuzy, A.; Zimmer, E.R.; Dubois, J.; Pruessner, J.; Cooperman, C.; Soucy, J.-P.; Kostikov, A.; Schirmaccher, E.; Désautels, R.; Gauthier, S.J.B.S.; et al. In vivo characterization of metabotropic glutamate receptor type 5 abnormalities in behavioral variant FTD. Brain Struct. Funct. 2016, 221, 1387–1402.
- Homayoun, H.; Moghaddam, B.J. Bursting of prefrontal cortex neurons in awake rats is regulated by metabotropic glutamate 5 (mGlu5) receptors: Rate-dependent influence and interaction with NMDA receptors. Cereb. Cortex 2005, 16, 93–105.
- Kalia, L.V.; Lang, A.E. Parkinson’s disease. Lancet 2015, 386, 896–912.
- Sveinbjornsdottir, S. The clinical symptoms of Parkinson’s disease. J. Neurochem. 2016, 13, 318–324.
- More, S.V.; Choi, D.-K. Emerging preclinical pharmacological targets for Parkinson’s disease. Oncotarget 2016, 7, 29835–29863.
- Aarsland, D.; Bronnick, K.; Williams-Gray, C.; Weintraub, D.; Marder, K.; Kulisevsky, J.; Burn, D.; Barone, P.; Pagonabarraga, J.; Allcock, L.; et al. Mild cognitive impairment in Parkinson disease: A multicenter pooled analysis. Neurology 2010, 75, 1062–1069.
- Hely, M.A.; Reid, W.G.; Adena, M.A.; Halliday, G.M.; Morris, J.G. The Sydney multicenter study of Parkinson’s disease: The inevitability of dementia at 20 years. Mov. Disord. 2008, 23, 837–844.
- Hisahara, S.; Shimohama, S. Dopamine Receptors and Parkinson’s Disease. Int. J. Med. Chem. 2011, 2011.
- Pezze, M.-A.; Dalley, J.W.; Robbins, T.W. Differential roles of dopamine D1 and D2 receptors in the nucleus accumbens in attentional performance on the five-choice serial reaction time task. Neuropsychopharmacol. Off. Publ. Am. Coll. Neuropsychopharmacol. 2007, 32, 273–283.
- Halliday, G.M.; Leverenz, J.B.; Schneider, J.S.; Adler, C.H. The neurobiological basis of cognitive impairment in Parkinson’s disease. Mov. Disord. Off. J. Mov. Disord. Soc. 2014, 29, 634–650.
- Ballard, C.; Ziabreva, I.; Perry, R.; Larsen, J.P.; O’Brien, J.; McKeith, I.; Perry, E.; Aarsland, D. Differences in neuropathologic characteristics across the Lewy body dementia spectrum. Neurology 2006, 67, 1931–1934.
- Shimada, H.; Hirano, S.; Shinotoh, H.; Aotsuka, A.; Sato, K.; Tanaka, N.; Ota, T.; Asahina, M.; Fukushi, K.; Kuwabara, S.; et al. Mapping of brain acetylcholinesterase alterations in Lewy body disease by PET. Neurology 2009, 73, 273–278.
- Varrone, A.; Svenningsson, P.; Marklund, P.; Fatouros-Bergman, H.; Forsberg, A.; Halldin, C.; Nilsson, L.G.; Farde, L. 5-HT1B receptor imaging and cognition: A positron emission tomography study in control subjects and Parkinson’s disease patients. Synapse 2015, 69, 365–374.
- Benedict, R.H.; Cookfair, D.; Gavett, R.; Gunther, M.; Munschauer, F.; Garg, N.; Weinstock-Guttman, B. Validity of the minimal assessment of cognitive function in multiple sclerosis (MACFIMS). J. Int. Neuropsychol. Soc. JINS 2006, 12, 549–558.
- Lubrini, G.; Perianez, J.A.; Rios-Lago, M.; Frank, A. Processing speed in relapsing-remitting multiple sclerosis: The role played by the depressive symptoms. Rev. Neurol. 2012, 55, 585–592.
- Arnett, P.A.; Higginson, C.I.; Voss, W.D.; Bender, W.I.; Wurst, J.M.; Tippin, J.M. Depression in multiple sclerosis: Relationship to working memory capacity. Neuropsychology 1999, 13, 546–556.
- Feinstein, A.; Magalhaes, S.; Richard, J.F.; Audet, B.; Moore, C. The link between multiple sclerosis and depression. Nat. Rev. Neurol. 2014, 10, 507–517.
- Ehde, D.M.; Kraft, G.H.; Chwastiak, L.; Sullivan, M.D.; Gibbons, L.E.; Bombardier, C.H.; Wadhwani, R. Efficacy of paroxetine in treating major depressive disorder in persons with multiple sclerosis. Gen. Hosp. Psychiatry 2008, 30, 40–48.
- Beal, M.F.; Ferrante, R.J.; Swartz, K.J.; Kowall, N.W. Chronic quinolinic acid lesions in rats closely resemble Huntington’s disease. J. Neurosci. Off. J. Soc. Neurosci. 1991, 11, 1649–1659.
- Bruno, V.; Ksiazek, I.; Battaglia, G.; Lukic, S.; Leonhardt, T.; Sauer, D.; Gasparini, F.; Kuhn, R.; Nicoletti, F.; Flor, P.J. Selective blockade of metabotropic glutamate receptor subtype 5 is neuroprotective. Neuropharmacology 2000, 39, 2223–2230.
- Schiefer, J.; Sprunken, A.; Puls, C.; Luesse, H.G.; Milkereit, A.; Milkereit, E.; Johann, V.; Kosinski, C.M. The metabotropic glutamate receptor 5 antagonist MPEP and the mGluR2 agonist LY379268 modify disease progression in a transgenic mouse model of Huntington’s disease. Brain Res. 2004, 1019, 246–254.
- Chen, J.Y.; Wang, E.A.; Cepeda, C.; Levine, M.S. Dopamine imbalance in Huntington’s disease: A mechanism for the lack of behavioral flexibility. Front. Neurosci. 2013, 7, 114.
- Charvin, D.; Vanhoutte, P.; Pages, C.; Borrelli, E.; Caboche, J. Unraveling a role for dopamine in Huntington’s disease: The dual role of reactive oxygen species and D2 receptor stimulation. Proc. Natl. Acad. Sci. USA 2005, 102, 12218–12223.
- Charvin, D.; Roze, E.; Perrin, V.; Deyts, C.; Betuing, S.; Pages, C.; Regulier, E.; Luthi-Carter, R.; Brouillet, E.; Deglon, N.; et al. Haloperidol protects striatal neurons from dysfunction induced by mutated huntingtin in vivo. Neurobiol. Dis. 2008, 29, 22–29.
- Meneses, A. 5-HT system and cognition. Neurosci. Biobehav. Rev. 1999, 23, 1111–1125.
- Buhot, M.C.; Martin, S.; Segu, L. Role of serotonin in memory impairment. Ann. Med. 2000, 32, 210–221.
- Codony, X.; Vela, J.M.; Ramírez, M.J. 5-HT6 receptor and cognition. Curr. Opin. Pharmacol. 2011, 11, 94–100.
- Fone, K.C. An update on the role of the 5-hydroxytryptamine6 receptor in cognitive function. Neuropharmacology 2008, 55, 1015–1022.
- Meneses, A.; Pérez-García, G.; Ponce-Lopez, T.; Castillo, C. 5-HT6 receptor memory and amnesia: Behavioral pharmacology–learning and memory processes. In International Review of Neurobiology; Elsevier: Amsterdam, The Netherlands, 2011; Volume 96, pp. 27–47.
- González-Vera, J.A.; Medina, R.A.; Martín-Fontecha, M.; Gonzalez, A.; de la Fuente, T.; Vázquez-Villa, H.; García-Cárceles, J.; Botta, J.; McCormick, P.J.; Benhamú, B.; et al. A new serotonin 5-HT6 receptor antagonist with procognitive activity—Importance of a halogen bond interaction to stabilize the binding. Sci. Rep. 2017, 7, 41293.
- Benhamú, B.; Martin-Fontecha, M.; Vázquez-Villa, H.; Pardo, L.; Lopez-Rodriguez, M.L.J. Serotonin 5-HT6 receptor antagonists for the treatment of cognitive deficiency in Alzheimer’s disease. J. Med. Chem. 2014, 57, 7160–7181.
- Ramírez, M.J. 5-HT 6 receptors and Alzheimer’s disease. Alzheimer’s Res. Ther. 2013, 5, 15.
- Marazziti, D.; Rutigliano, G.; Catena-Dell’Osso, M.; Baroni, S.; Dell’Osso, L. The 5-HT6 receptor antagonism approach in Alzheimer’s disease. Drugs Future 2014, 39, 133–140.
- Mohler, E.G.; Baker, P.M.; Gannon, K.S.; Jones, S.S.; Shacham, S.; Sweeney, J.A.; Ragozzino, M.E. The effects of PRX-07034, a novel 5-HT6 antagonist, on cognitive flexibility and working memory in rats. Psychopharmacology 2012, 220, 687–696.
- Ferrero, H.; Solas, M.; Francis, P.T.; Ramirez, M.J. Serotonin 5-HT6 Receptor Antagonists in Alzheimer’s Disease: Therapeutic Rationale and Current Development Status. CNS Drugs 2017, 31, 19–32.
- Andrews, M.; Tousi, B.; Sabbagh, M.N. 5HT6 Antagonists in the Treatment of Alzheimer’s Dementia: Current Progress. Neurol. Ther. 2018, 7, 51–58.
- Halford, J.C.; Harrold, J.A.; Boyland, E.J.; Lawton, C.L.; Blundell, J.E. Serotonergic drugs: Effects on appetite expression and use for the treatment of obesity. Drugs 2007, 67, 27–55.
- Ivachtchenko, A.V.; Ivanenkov, Y.A.; Veselov, M.S.; Okun, I.M. AVN-322 is a Safe Orally Bio-Available Potent and Highly Selective Antagonist of 5-HT6R with Demonstrated Ability to Improve Impaired Memory in Animal Models. Curr. Alzheimer Res. 2017, 14, 268–294.
- Ivachtchenko, A.V.; Lavrovsky, Y.; Okun, I. AVN-101: A Multi-Target Drug Candidate for the Treatment of CNS Disorders. J. Alzheimers Dis. JAD 2016, 53, 583–620.
- Ivachtchenko, A.V.; Lavrovsky, Y.; Ivanenkov, Y.A. AVN-211, Novel and Highly Selective 5-HT6 Receptor Small Molecule Antagonist, for the Treatment of Alzheimer’s Disease. Mol. Pharm. 2016, 13, 945–963.
- Schneider, L.S. Idalopirdine for Alzheimer’s disease: Written in the stars. Lancet Neurol. 2014, 13, 1063–1065.
- Roman, G.C.; Wilkinson, D.G.; Doody, R.S.; Black, S.E.; Salloway, S.P.; Schindler, R.J. Donepezil in vascular dementia: Combined analysis of two large-scale clinical trials. Dement. Geriatr. Cogn. Disord. 2005, 20, 338–344.
- Maher-Edwards, G.; Zvartau-Hind, M.; Hunter, A.J.; Gold, M.; Hopton, G.; Jacobs, G.; Davy, M.; Williams, P. Double-blind, controlled phase II study of a 5-HT6 receptor antagonist, SB-742457, in Alzheimer’s disease. Curr. Alzheimer Res. 2010, 7, 374–385.
- Maher-Edwards, G.; Dixon, R.; Hunter, J.; Gold, M.; Hopton, G.; Jacobs, G.; Hunter, J.; Williams, P. SB-742457 and donepezil in Alzheimer disease: A randomized, placebo-controlled study. Int. J. Geriatr. Psychiatry 2011, 26, 536–544.
- Maher-Edwards, G.; Watson, C.; Ascher, J.; Barnett, C.; Boswell, D.; Davies, J.; Fernandez, M.; Kurz, A.; Zanetti, O.; Safirstein, B.; et al. Two randomized controlled trials of SB742457 in mild-to-moderate Alzheimer’s disease. Alzheimers Dement. 2015, 1, 23–36.
- Doody, R.S.; Gavrilova, S.I.; Sano, M.; Thomas, R.G.; Aisen, P.S.; Bachurin, S.O.; Seely, L.; Hung, D. Effect of dimebon on cognition, activities of daily living, behaviour, and global function in patients with mild-to-moderate Alzheimer’s disease: A randomised, double-blind, placebo-controlled study. Lancet 2008, 372, 207–215.
- Nirogi, R.; Kandikere, V.; Mudigonda, K.; Bhyrapuneni, G.; Jasti, V. Safety, tolerability and pharmacokinetics of SUVN-502—A 5-HT6 receptor antagonist, first in human phase-1 Single Ascending Dose (SAD) study. Alzheimers Dement. J. Alzheimers Assoc. 2009, 5, P250.
- Nirogi, R.; Ajjala, D.R.; Aleti, R.; Rayapati, L.; Pantangi, H.R.; Boggavarapu, R.K.; Padala, N.S.P. Development and validation of sensitive LC-MS/MS method for the quantification of SUVN-502 and its metabolite and its application for first in human pharmacokinetic study. J. Pharm. Biomed. Anal. 2017, 145, 423–430.
- Nirogi, R.; Shinde, A.; Kambhampati, R.S.; Mohammed, A.R.; Saraf, S.K.; Badange, R.K.; Bandyala, T.R.; Bhatta, V.; Bojja, K.; Reballi, V.J. Discovery and Development of 1-[(2-Bromophenyl) sulfonyl]-5-methoxy-3-[(4-methyl-1-piperazinyl) methyl]-1 H-indole Dimesylate Monohydrate (SUVN-502): A Novel, Potent, Selective and Orally Active Serotonin 6 (5-HT6) Receptor Antagonist for Potential Treatment of Alzheimer’s Disease. J. Med. Chem. 2017, 60, 1843–1859.
- Callegari, I.; Mattei, C.; Benassi, F.; Krueger, F.; Grafman, J.; Yaldizli, Ö.; Sassos, D.; Massucco, D.; Scialò, C.; Nobili, F. Agomelatine improves apathy in frontotemporal dementia. Neurodegener. Dis. 2016, 16, 352–356.
- Karaiskos, D.; Pappa, D.; Katirtzoglou, E.; Siarkos, K.; Tzavellas, E.; Papadimitriou, G.; Politis, A. EPA-1197–Agomelatine for treating apathy in alzheimer’s disease. Eur. Psychiatry 2014, 29, 1.
- Avila, A.; Cardona, X.; Martin-Baranera, M.; Leon, L.; Caballol, N.; Millet, P.; Bello, J.J. Agomelatine for depression in Parkinson disease: Additional effect on sleep and motor dysfunction. J. Clin. Psychopharmacol. 2015, 35, 719–723.
- Biskup, C.S.; Sanchez, C.L.; Arrant, A.; Van Swearingen, A.E.; Kuhn, C.; Zepf, F.D. Effects of acute tryptophan depletion on brain serotonin function and concentrations of dopamine and norepinephrine in C57BL/6J and BALB/cJ mice. PLoS ONE 2012, 7, e35916.
- Jans, L.A.; Korte-Bouws, G.A.; Korte, S.M.; Blokland, A. The effects of acute tryptophan depletion on affective behaviour and cognition in Brown Norway and Sprague Dawley rats. J. Psychopharmacol. 2010, 24, 605–614.
- Young, S.N.; Ervin, F.R.; Pihl, R.O.; Finn, P. Biochemical aspects of tryptophan depletion in primates. Psychopharmacology 1989, 98, 508–511.
- Riedel, W.J.; Klaassen, T.; Deutz, N.E.; van Someren, A.; van Praag, H.M. Tryptophan depletion in normal volunteers produces selective impairment in memory consolidation. Psychopharmacology 1999, 141, 362–369.
- Mendelsohn, D.; Riedel, W.J.; Sambeth, A. Effects of acute tryptophan depletion on memory, attention and executive functions: A systematic review. Neurosci. Biobehav. Rev. 2009, 33, 926–952.
- Lash, T.L.; Silliman, R.A.; Guadagnoli, E.; Mor, V. The effect of less than definitive care on breast carcinoma recurrence and mortality. Cancer 2000, 89, 1739–1747.
- Mohajeri, M.H.; Wittwer, J.; Vargas, K.; Hogan, E.; Holmes, A.; Rogers, P.J.; Goralczyk, R.; Gibson, E.L. Chronic treatment with a tryptophan-rich protein hydrolysate improves emotional processing, mental energy levels and reaction time in middle-aged women. Br. J. Nutr. 2015, 113, 350–365.
- Rondanelli, M.; Opizzi, A.; Faliva, M.; Mozzoni, M.; Antoniello, N.; Cazzola, R.; Savare, R.; Cerutti, R.; Grossi, E.; Cestaro, B. Effects of a diet integration with an oily emulsion of DHA-phospholipids containing melatonin and tryptophan in elderly patients suffering from mild cognitive impairment. Nutr. Neurosci. 2012, 15, 46–54.
- Pae, C.-U.; Wang, S.-M.; Han, C.; Lee, S.-J.; Patkar, A.A.; Masand, P.S.; Serretti, A. Vortioxetine: A meta-analysis of 12 short-term, randomized, placebo-controlled clinical trials for the treatment of major depressive disorder. J. Psychiatry Neurosci. 2015, 40, 174–186.
- Almeida, S.; Glahn, D.C.; Argyropoulos, S.V.; Frangou, S. Acute citalopram administration may disrupt contextual information processing in healthy males. Eur. Psychiatry J. Assoc. Eur. Psychiatr. 2010, 25, 87–91.
- Yu, Y.; Patch, C.; Weston-Green, K.; Zhou, Y.; Zheng, K.; Huang, X.F. Dietary Galacto-Oligosaccharides and Resistant Starch Protect Against Altered CB1 and 5-HT1A and 2A Receptor Densities in Rat Brain: Implications for Preventing Cognitive and Appetite Dysfunction During a High-Fat Diet. Mol. Nutr. Food Res. 2018, 62, e1800422.
- Carlsson, A. The occurrence, distribution and physiological role of catecholamines in the nervous system. Pharmacol. Rev. 1959, 11, 490–493.
- Girault, J.-A.; Greengard, P. The Neurobiology of Dopamine Signaling. Arch. Neurol. 2004, 61, 641–644.
- Westbrook, A.; Braver, T.S. Dopamine Does Double Duty in Motivating Cognitive Effort. Neuron 2016, 89, 695–710.
- Grace, A.A. Dysregulation of the dopamine system in the pathophysiology of schizophrenia and depression. Nat. Rev. Neurosci. 2016, 17, 524–532.
- Arnsten, A.F.; Wang, M.; Paspalas, C.D. Dopamine’s Actions in Primate Prefrontal Cortex: Challenges for Treating Cognitive Disorders. Pharmacol. Rev. 2015, 67, 681–696.
- Blum, K.; Badgaiyan, R.D.; Dunston, G.M.; Baron, D.; Modestino, E.J.; McLaughlin, T.; Steinberg, B.; Gold, M.S.; Gondre-Lewis, M.C. The DRD2 Taq1A A1 Allele May Magnify the Risk of Alzheimer’s in Aging African-Americans. Mol. Neurobiol. 2018, 55, 5526–5536.
- Karthivashan, G.; Ganesan, P.; Park, S.-Y.; Lee, H.-W.; Choi, D.-K. Lipid-based nanodelivery approaches for dopamine-replacement therapies in Parkinson’s disease: From preclinical to translational studies. Biomaterials 2020, 232, 119704.
- van Wimersma Greidanus, T.B.; Jolles, J.; De Wied, D. Hypothalamic neuropeptides and memory. Acta Neurochir. 1985, 75, 99–105.
- Fallon, S.J.; Muhammed, K.; Drew, D.S.; Ang, Y.-S.; Manohar, S.G.; Husain, M. Dopamine guides competition for cognitive control: Common effects of haloperidol on working memory and response conflict. Cortex 2019, 113, 156–168.
- Kapur, S.; Seeman, P. Does fast dissociation from the dopamine D2 receptor explain the action of atypical antipsychotics? A new hypothesis. Am. J. Psychiatry 2001, 158, 360–369.
- Marshall, C.A.; Brodnik, Z.D.; Mortensen, O.V.; Reith, M.E.A.; Shumsky, J.S.; Waterhouse, B.D.; España, R.A.; Kortagere, S. Selective activation of Dopamine D3 receptors and norepinephrine transporter blockade enhances sustained attention. Neuropharmacology 2019, 148, 178–188.
- Morena, M.; Campolongo, P. The endocannabinoid system: An emotional buffer in the modulation of memory function. Neurobiol. Learn. Mem. 2014, 112, 30–43.
- Campolongo, P.; Ratano, P.; Manduca, A.; Scattoni, M.L.; Palmery, M.; Trezza, V.; Cuomo, V. The endocannabinoid transport inhibitor AM404 differentially modulates recognition memory in rats depending on environmental aversiveness. Front. Behav. Neurosci. 2012, 6.
- Campolongo, P.; Roozendaal, B.; Trezza, V.; Hauer, D.; Schelling, G.; McGaugh, J.L.; Cuomo, V. Endocannabinoids in the rat basolateral amygdala enhance memory consolidation and enable glucocorticoid modulation of memory. Proc. Natl. Acad. Sci. USA 2009, 106, 4888–4893.
- Ferland, J.-M.N.; Carr, M.R.; Lee, A.M.; Hoogeland, M.E.; Winstanley, C.A.; Pattij, T. Examination of the effects of cannabinoid ligands on decision making in a rat gambling task. Pharmacol. Biochem. Behav. 2018, 170, 87–97.
- Irimia, C.; Polis, I.Y.; Stouffer, D.; Parsons, L.H. Persistent effects of chronic Δ9-THC exposure on motor impulsivity in rats. Psychopharmacology 2015, 232, 3033–3043.
- Milton, N.G. Anandamide and noladin ether prevent neurotoxicity of the human amyloid-beta peptide. Neurosci. Lett. 2002, 332, 127–130.
- Mazzola, C.; Micale, V.; Drago, F. Amnesia induced by beta-amyloid fragments is counteracted by cannabinoid CB1 receptor blockade. Eur. J. Pharmacol. 2003, 477, 219–225.
- Manuel, I.; Lombardero, L.; LaFerla, F.M.; Gimenez-Llort, L.; Rodriguez-Puertas, R. Activity of muscarinic, galanin and cannabinoid receptors in the prodromal and advanced stages in the triple transgenic mice model of Alzheimer’s disease. Neuroscience 2016, 329, 284–293.
- Fakhfouri, G.; Ahmadiani, A.; Rahimian, R.; Grolla, A.A.; Moradi, F.; Haeri, A. WIN55212-2 attenuates amyloid-beta-induced neuroinflammation in rats through activation of cannabinoid receptors and PPAR-gamma pathway. Neuropharmacology 2012, 63, 653–666.
- Tolon, R.M.; Nunez, E.; Pazos, M.R.; Benito, C.; Castillo, A.I.; Martinez-Orgado, J.A.; Romero, J. The activation of cannabinoid CB2 receptors stimulates in situ and in vitro beta-amyloid removal by human macrophages. Brain Res. 2009, 1283, 148–154.
- Wu, J.; Bie, B.; Yang, H.; Xu, J.J.; Brown, D.L.; Naguib, M. Activation of the CB2 receptor system reverses amyloid-induced memory deficiency. Neurobiol. Aging 2013, 34, 791–804.
- Fernandez-Ruiz, J.; Romero, J.; Velasco, G.; Tolon, R.M.; Ramos, J.A.; Guzman, M. Cannabinoid CB2 receptor: A new target for controlling neural cell survival? Trends Pharmacol. Sci. 2007, 28, 39–45.
- Sagredo, O.; Gonzalez, S.; Aroyo, I.; Pazos, M.R.; Benito, C.; Lastres-Becker, I.; Romero, J.P.; Tolon, R.M.; Mechoulam, R.; Brouillet, E.; et al. Cannabinoid CB2 receptor agonists protect the striatum against malonate toxicity: Relevance for Huntington’s disease. Glia 2009, 57, 1154–1167.
- Palazuelos, J.; Aguado, T.; Pazos, M.R.; Julien, B.; Carrasco, C.; Resel, E.; Sagredo, O.; Benito, C.; Romero, J.; Azcoitia, I.; et al. Microglial CB2 cannabinoid receptors are neuroprotective in Huntington’s disease excitotoxicity. Brain J. Neurol. 2009, 132, 3152–3164.
- Lastres-Becker, I.; Bizat, N.; Boyer, F.; Hantraye, P.; Brouillet, E.; Fernandez-Ruiz, J. Effects of cannabinoids in the rat model of Huntington’s disease generated by an intrastriatal injection of malonate. Neuroreport 2003, 14, 813–816.
- Laprairie, R.B.; Bagher, A.M.; Kelly, M.E.; Denovan-Wright, E.M. Cannabidiol is a negative allosteric modulator of the cannabinoid CB1 receptor. Br. J. Pharmacol. 2015, 172, 4790–4805.
- Laprairie, R.B.; Bagher, A.M.; Kelly, M.E.; Denovan-Wright, E.M. Biased Type 1 Cannabinoid Receptor Signaling Influences Neuronal Viability in a Cell Culture Model of Huntington Disease. Mol. Pharmacol. 2016, 89, 364–375.
- Diaz-Alonso, J.; Paraiso-Luna, J.; Navarrete, C.; Del Rio, C.; Cantarero, I.; Palomares, B.; Aguareles, J.; Fernandez-Ruiz, J.; Bellido, M.L.; Pollastro, F.; et al. VCE-003.2, a novel cannabigerol derivative, enhances neuronal progenitor cell survival and alleviates symptomatology in murine models of Huntington’s disease. Sci. Rep. 2016, 6, 29789.
- Rodriguez-Cueto, C.; Santos-Garcia, I.; Garcia-Toscano, L.; Espejo-Porras, F.; Bellido, M.; Fernandez-Ruiz, J.; Munoz, E.; de Lago, E. Neuroprotective effects of the cannabigerol quinone derivative VCE-003.2 in SOD1(G93A) transgenic mice, an experimental model of amyotrophic lateral sclerosis. Biochem. Pharmacol. 2018, 157, 217–226.
- Colangelo, C.; Shichkova, P.; Keller, D.; Markram, H.; Ramaswamy, S. Cellular, Synaptic and Network Effects of Acetylcholine in the Neocortex. Front. Neural Circuits 2019, 13.
- Wevers, A. Localisation of pre- and postsynaptic cholinergic markers in the human brain. Behav. Brain Res. 2011, 221, 341–355.
- Dani, J.A. Overview of nicotinic receptors and their roles in the central nervous system. Biol. Psychiatry 2001, 49, 166–174.
- Quik, M.; Bordia, T.; O’Leary, K. Nicotinic receptors as CNS targets for Parkinson’s disease. Biochem. Pharmacol. 2007, 74, 1224–1234.
- Quik, M.; Wonnacott, S. α6β2* and α4β2* Nicotinic Acetylcholine Receptors as Drug Targets for Parkinson’s Disease. J. Pharmacol. Rev. 2011, 63, 938–966.
- Nelson, A.B.; Bussert, T.G.; Kreitzer, A.C.; Seal, R.P. Striatal Cholinergic Neurotransmission Requires VGLUT3. J. Neurosci. 2014, 34, 8772–8777.
- English, D.F.; Ibanez-Sandoval, O.; Stark, E.; Tecuapetla, F.; Buzsáki, G.; Deisseroth, K.; Tepper, J.M.; Koos, T. GABAergic circuits mediate the reinforcement-related signals of striatal cholinergic interneurons. Nat. Neurosci. 2011, 15, 123–130.
- Rice, M.E.; Patel, J.C.; Cragg, S.J. Dopamine release in the basal ganglia. Neuroscience 2011, 198, 112–137.
- Zhou, F.-M.; Liang, Y.; Dani, J.A. Endogenous nicotinic cholinergic activity regulates dopamine release in the striatum. Nat. Neurosci. 2001, 4, 1224–1229.
- Liu, Y.; Hu, J.; Wu, J.; Zhu, C.; Hui, Y.; Han, Y.; Huang, Z.; Ellsworth, K.; Fan, W. α7 nicotinic acetylcholine receptor-mediated neuroprotection against dopaminergic neuron loss in an MPTP mouse model via inhibition of astrocyte activation. J. Neuroinflammation 2012, 9, 98.
- Foucault-Fruchard, L.; Doméné, A.; Page, G.; Windsor, M.; Emond, P.; Rodrigues, N.; Dollé, F.; Damont, A.; Buron, F.; Routier, S.; et al. Neuroprotective effect of the alpha 7 nicotinic receptor agonist PHA 543613 in an in vivo excitotoxic adult rat model. Neuroscience 2017, 356, 52–63.
- Medeiros, R.; Castello, N.A.; Cheng, D.; Kitazawa, M.; Baglietto-Vargas, D.; Green, K.N.; Esbenshade, T.A.; Bitner, R.S.; Decker, M.W.; LaFerla, F.M. alpha7 Nicotinic receptor agonist enhances cognition in aged 3xTg-AD mice with robust plaques and tangles. Am. J. Pathol. 2014, 184, 520–529.
- Vicens, P.; Heredia, L.; Torrente, M.; Domingo, J.L. Behavioural effects of PNU-282987 and stress in an animal model of Alzheimer’s disease. Psychogeriatr. Off. J. Jpn. Psychogeriatr. Soc. 2017, 17, 33–42.
- Van Kampen, M.; Selbach, K.; Schneider, R.; Schiegel, E.; Boess, F.; Schreiber, R. AR-R 17779 improves social recognition in rats by activation of nicotinic alpha7 receptors. Psychopharmacology 2004, 172, 375–383.
- Boess, F.G.; De Vry, J.; Erb, C.; Flessner, T.; Hendrix, M.; Luithle, J.; Methfessel, C.; Riedl, B.; Schnizler, K.; van der Staay, F.J.; et al. The novel alpha7 nicotinic acetylcholine receptor agonist N-[(3R)-1-azabicyclo[2.2.2]oct-3-yl]-7-[2-(methoxy)phenyl]-1-benzofuran-2-carboxa mide improves working and recognition memory in rodents. J. Pharmacol. Exp. Ther. 2007, 321, 716–725.
- Lahmy, V.; Long, R.; Morin, D.; Villard, V.; Maurice, T. Mitochondrial protection by the mixed muscarinic/sigma1 ligand ANAVEX2-73, a tetrahydrofuran derivative, in Abeta25-35 peptide-injected mice, a nontransgenic Alzheimer’s disease model. Front. Cell. Neurosci. 2014, 8, 463.
- Nicoletti, F.; Bockaert, J.; Collingridge, G.L.; Conn, P.J.; Ferraguti, F.; Schoepp, D.D.; Wroblewski, J.T.; Pin, J.P. Metabotropic glutamate receptors: From the workbench to the bedside. Neuropharmacology 2011, 60, 1017–1041.
- Niswender, C.M.; Conn, P.J. Metabotropic glutamate receptors: Physiology, pharmacology, and disease. Annu. Rev. Pharmacol. Toxicol. 2010, 50, 295–322.
- Iyer, A.M.; van Scheppingen, J.; Milenkovic, I.; Anink, J.J.; Lim, D.; Genazzani, A.A.; Adle-Biassette, H.; Kovacs, G.G.; Aronica, E. Metabotropic glutamate receptor 5 in Down’s syndrome hippocampus during development: Increased expression in astrocytes. Curr. Alzheimer Res. 2014, 11, 694–705.
- Spampinato, S.F.; Copani, A.; Nicoletti, F.; Sortino, M.A.; Caraci, F. Metabotropic Glutamate Receptors in Glial Cells: A New Potential Target for Neuroprotection? Front. Mol. Neurosci. 2018, 11, 414.
- Chen, X.; Lin, R.; Chang, L.; Xu, S.; Wei, X.; Zhang, J.; Wang, C.; Anwyl, R.; Wang, Q. Enhancement of long-term depression by soluble amyloid β protein in rat hippocampus is mediated by metabotropic glutamate receptor and involves activation of p38MAPK, STEP and caspase-3. Neuroscience 2013, 253, 435–443.
- Renner, M.; Lacor, P.N.; Velasco, P.T.; Xu, J.; Contractor, A.; Klein, W.L.; Triller, A. Deleterious Effects of Amyloid β Oligomers Acting as an Extracellular Scaffold for mGluR5. Neuron 2010, 66, 739–754.
- Caraci, F.; Molinaro, G.; Battaglia, G.; Giuffrida, M.L.; Riozzi, B.; Traficante, A.; Bruno, V.; Cannella, M.; Merlo, S.; Wang, X.; et al. Targeting Group II Metabotropic Glutamate (mGlu) Receptors for the Treatment of Psychosis Associated with Alzheimer’s Disease: Selective Activation of mGlu2 Receptors Amplifies β-Amyloid Toxicity in Cultured Neurons, Whereas Dual Activation of mGlu2 and mGlu3 Receptors Is Neuroprotective. Mol. Pharmacol. 2011, 79, 618–626.
- Tang, X.-L.; Wang, Y.; Li, D.-L.; Luo, J.; Liu, M.-Y. Orphan G protein-coupled receptors (GPCRs): Biological functions and potential drug targets. Acta Pharmacol. Sin. 2012, 33, 363–371.
- Grottick, A.; Schuelert, N.; Rosenbrock, H.; von Heimendahl, M.; Arban, R.; Hobson, S.J.B.P. 669. GPR52 Agonists Represent a Novel Approach to Treat Cognitive Deficits Associated with Schizophrenia. Boil. Psychiatry 2017, 81, S271.
- Haque, M.E.; Kim, I.-S.; Jakaria, M.; Akther, M.; Choi, D.-K. Importance of GPCR-Mediated Microglial Activation in Alzheimer’s Disease. Front. Cell. Neurosci. 2018, 12, 12.
- Huang, Y.; Skwarek-Maruszewska, A.; Horré, K.; Vandewyer, E.; Wolfs, L.; Snellinx, A.; Saito, T.; Radaelli, E.; Corthout, N.; Colombelli, J. Loss of GPR3 reduces the amyloid plaque burden and improves memory in Alzheimer’s disease mouse models. Sci. Transl. Med. 2015, 7, 309ra164.
- Khrimian, L.; Obri, A.; Ramos-Brossier, M.; Rousseaud, A.; Moriceau, S.; Nicot, A.-S.; Mera, P.; Kosmidis, S.; Karnavas, T.; Saudou, F.J. Gpr158 mediates osteocalcin’s regulation of cognition. J. Exp. Med. 2017, 214, 2859–2873.
- Ehrlich, A.T.; Maroteaux, G.; Robe, A.; Venteo, L.; Nasseef, M.T.; van Kempen, L.C.; Mechawar, N.; Turecki, G.; Darcq, E.; Kieffer, B.L.J.C.b. Expression map of 78 brain-expressed mouse orphan GPCRs provides a translational resource for neuropsychiatric research. Commun. Biol. 2018, 1, 102.
- Thathiah, A.; Spittaels, K.; Hoffmann, M.; Staes, M.; Cohen, A.; Horre, K.; Vanbrabant, M.; Coun, F.; Baekelandt, V.; Delacourte, A.; et al. The orphan G protein-coupled receptor 3 modulates amyloid-beta peptide generation in neurons. Science 2009, 323, 946–951.
- Sutton, L.P.; Orlandi, C.; Song, C.; Oh, W.C.; Muntean, B.S.; Xie, K.; Filippini, A.; Xie, X.; Satterfield, R.; Yaeger, J.D. Orphan receptor GPR158 controls stress-induced depression. eLife 2018, 7, e33273.
- Komatsu, H.; Maruyama, M.; Yao, S.; Shinohara, T.; Sakuma, K.; Imaichi, S.; Chikatsu, T.; Kuniyeda, K.; Siu, F.K.; Peng, L.S. Anatomical transcriptome of G protein-coupled receptors leads to the identification of a novel therapeutic candidate GPR52 for psychiatric disorders. PLoS ONE 2014, 9, e90134.
- Valverde, O.; Celerier, E.; Baranyi, M.; Vanderhaeghen, P.; Maldonado, R.; Sperlagh, B.; Vassart, G.; Ledent, C. GPR3 receptor, a novel actor in the emotional-like responses. PLoS ONE 2009, 4, e4704.
- Hurst, K.; Badgley, C.; Ellsworth, T.; Bell, S.; Friend, L.; Prince, B.; Welch, J.; Cowan, Z.; Williamson, R.; Lyon, C. A putative lysophosphatidylinositol receptor GPR55 modulates hippocampal synaptic plasticity. Hippocampus 2017, 27, 985–998.
- Matsumoto, M.; Saito, T.; Takasaki, J.; Kamohara, M.; Sugimoto, T.; Kobayashi, M.; Tadokoro, M.; Matsumoto, S.-I.; Ohishi, T.; Furuichi, K.J.B.; et al. An evolutionarily conserved G-protein coupled receptor family, SREB, expressed in the central nervous system. Biochem. Biophys. Res. Commun. 2000, 272, 576–582.
- Yanai, T.; Kurosawa, A.; Nikaido, Y.; Nakajima, N.; Saito, T.; Osada, H.; Konno, A.; Hirai, H.; Takeda, S. Identification and molecular docking studies for novel inverse agonists of SREB, super conserved receptor expressed in brain. Genes Cells 2016, 21, 717–727.
- Hellebrand, S.; Wittenberger, T.; Schaller, H.C.; Hermans-Borgmeyer, I. Gpr85, a novel member of the G-protein coupled receptor family, prominently expressed in the developing mouse cerebral cortex. Gene Expr. Patterns 2001, 1, 13–16.
More
Information
Subjects:
Neurosciences
Contributor
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
1.7K
Entry Collection:
Neurodegeneration
Revisions:
2 times
(View History)
Update Date:
20 May 2021
Table of Contents



Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No