 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Oscar Campuzano | + 1613 word(s) | 1613 | 2021-06-25 10:05:53 | | | |
| 2 | Lily Guo | Meta information modification | 1613 | 2021-06-28 02:44:55 | | |
Video Upload Options
Congenital heart disease is a group of pathologies characterized by structural malformations of the heart or great vessels. These alterations occur during the embryonic period and are the most frequently observed severe congenital malformations, the main cause of neonatal mortality due to malformation, and the second most frequent congenital malformations overall after malformations of the central nervous system. The severity of different types of congenital heart disease varies depending on the combination of associated anatomical defects. The causes of these malformations are usually considered multifactorial, but genetic variants play a key role. Currently, use of high-throughput genetic technologies allows identification of pathogenic aneuploidies, deletions/duplications of large segments, as well as rare single nucleotide variants. The high incidence of congenital heart disease as well as the associated complications makes it necessary to establish a diagnosis as early as possible to adopt the most appropriate measures in a personalized approach. In this review, we provide an exhaustive update of the genetic bases of the most frequent congenital heart diseases as well as other syndromes associated with congenital heart defects, and how genetic data can be translated to clinical practice in a personalized approach.
1. Introduction
2. Epidemiology
3. Genetic Basis
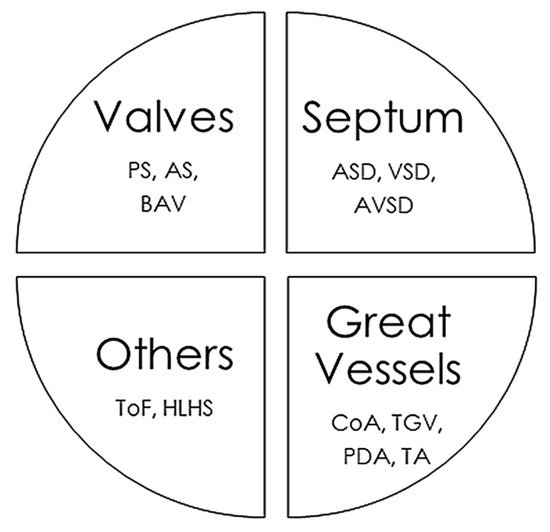
4. Cardiac Malformations
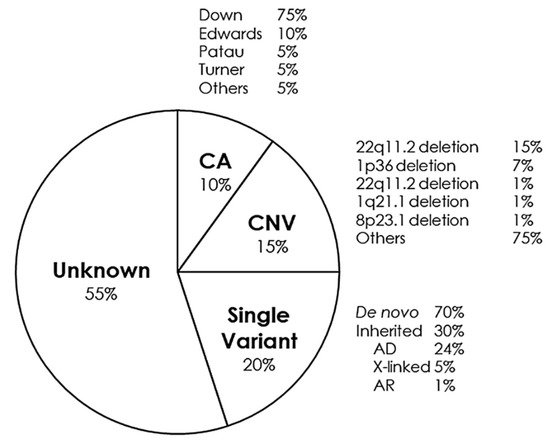
5. Syndromes with Congenital Cardiac Alteration
6. Conclusions
References
- Campbell, M. Genetic and environmental factors in congenital heart disease. Q. J. Med. 1949, 18, 379–391.
- Nora, J.J. Multifactorial inheritance hypothesis for the etiology of congenital heart diseases. The genetic-environmental interaction. Circulation 1968, 38, 604–617.
- Shi, H.; O’Reilly, V.C.; Moreau, J.L.; Bewes, T.R.; Yam, M.X.; Chapman, B.E.; Grieve, S.M.; Stocker, R.; Graham, R.M.; Chapman, G.; et al. Gestational stress induces the unfolded protein response, resulting in heart defects. Development 2016, 143, 2561–2572.
- Vecoli, C.; Pulignani, S.; Foffa, I.; Andreassi, M.G. Congenital heart disease. the crossroads of genetics, epigenetics and environment. Curr. Genomics 2014, 15, 390–399.
- De Backer, J.; Callewaert, B.; Muino Mosquera, L. Genetics in congenital heart disease. Are we ready for it? Rev. Esp. Cardiol. 2020, 73, 937–947.
- Leirgul, E.; Fomina, T.; Brodwall, K.; Greve, G.; Holmstrom, H.; Vollset, S.E.; Tell, G.S.; Oyen, N. Birth prevalence of congenital heart defects in Norway 1994-2009—A nationwide study. Am. Heart J. 2014, 168, 956–964.
- Bernier, P.L.; Stefanescu, A.; Samoukovic, G.; Tchervenkov, C.I. The challenge of congenital heart disease worldwide. epidemiologic and demographic facts. Semin. Thorac. Cardiovasc. Surg. Pediatr. Card. Surg. Annu. 2010, 13, 26–34.
- Perez-Lescure Picarzo, J.; Mosquera Gonzalez, M.; Latasa Zamalloa, P.; Crespo Marcos, D. Incidence and evolution of congenital heart disease in Spain from 2003 until 2012. An. Pediatr. 2018, 89, 294–301.
- Jacobs, J.P.; Mayer, J.E., Jr.; Pasquali, S.K.; Hill, K.D.; Overman, D.M.; St Louis, J.D.; Kumar, S.R.; Backer, C.L.; Tweddell, J.S.; Dearani, J.A.; et al. The Society of Thoracic Surgeons Congenital Heart Surgery database. 2019 update on outcomes and quality. Ann. Thorac. Surg. 2019, 107, 691–704.
- Jacobs, M.L.; Jacobs, J.P.; Hill, K.D.; O’Brien, S.M.; Pasquali, S.K.; Vener, D.; Kumar, S.R.; Chiswell, K.; St Louis, J.D.; Mayer, J.E.; et al. The Society of Thoracic Surgeons Congenital Heart Surgery database. 2019 update on research. Ann. Thorac. Surg. 2019, 108, 671–679.
- Raissadati, A.; Nieminen, H.; Jokinen, E.; Sairanen, H. Progress in late results among pediatric cardiac surgery patients. a population-based 6-decade study with 98% follow-up. Circulation 2015, 131, 347–353.
- Gilboa, S.M.; Mai, C.T.; Shapiro-Mendoza, C.K.; Cragan, J.D.; Moore, C.A.; Meaney-Delman, D.M.; Jamieson, D.J.; Honein, M.A.; Boyle, C.A. Population-based pregnancy and birth defects surveillance in the era of Zika virus. Birth Defects Res. 2017, 109, 372–378.
- Pierpont, M.E.; Brueckner, M.; Chung, W.K.; Garg, V.; Lacro, R.V.; McGuire, A.L.; Mital, S.; Priest, J.R.; Pu, W.T.; Roberts, A.; et al. Genetic basis for congenital heart disease. revisited. A scientific statement from the American Heart Association. Circulation 2018, 138, e653–e711.
- Zaidi, S.; Brueckner, M. Genetics and genomics of congenital heart disease. Circ. Res. 2017, 120, 923–940.
- Nees, S.N.; Chung, W.K. The genetics of isolated congenital heart disease. Am. J. Med. Genet. C Semin. Med. Genet. 2020, 184, 97–106.
- Marin-Garcia, J. Advances in molecular genetics of congenital heart disease. Rev. Esp. Cardiol. 2009, 62, 242–245.


