Congenital heart disease is a group of pathologies characterized by structural malformations of the heart or great vessels. These alterations occur during the embryonic period and are the most frequently observed severe congenital malformations, the main cause of neonatal mortality due to malformation, and the second most frequent congenital malformations overall after malformations of the central nervous system. The severity of different types of congenital heart disease varies depending on the combination of associated anatomical defects. The causes of these malformations are usually considered multifactorial, but genetic variants play a key role. Currently, use of high-throughput genetic technologies allows identification of pathogenic aneuploidies, deletions/duplications of large segments, as well as rare single nucleotide variants. The high incidence of congenital heart disease as well as the associated complications makes it necessary to establish a diagnosis as early as possible to adopt the most appropriate measures in a personalized approach. In this review, we provide an exhaustive update of the genetic bases of the most frequent congenital heart diseases as well as other syndromes associated with congenital heart defects, and how genetic data can be translated to clinical practice in a personalized approach.
1. Introduction
Congenital cardiac alterations are a heterogeneous group of structural heart or great vessel malformations that arise during embryogenesis, usually between the third and tenth week of gestation. These types of defects give rise to congenital heart disease (CHD). The first report focused on CHD origin was published in 1950
[1], and multifactorial inheritance was hypothesized
[2]. Now, CHD has a known multifactorial etiology combining genetic alterations and environmental factors
[3][4].
Despite this complex origin, unraveling the potential cause of the disease will help to prevent new cases in future gestations. Therefore, a proper genetic diagnosis of CHD is crucial in personalized medicine. Recently, improvement of high throughput genetic techniques has allowed cost-effective genetic diagnosis; identification of the genetic alterations responsible for CHDs facilitates counseling regarding the recurrence risk in relatives and descendants, and prenatal and preimplantation genetic diagnosis may influence reproductive options
[5]. The technical approach should be chosen according to the suspected genetic alteration and based on the clinical diagnosis. For example, if Down syndrome is suspected, the appropriate genetic approach is a karyotype, not a complete exome or genome. It is important to note that the diagnostic performance of the genetic analysis is not always successful, so that some cases remain without a definitive genetic diagnosis after the study is carried out. This situation usually occurs when a genetic alteration is identified but the interpretation is not conclusive, leaving an ambiguous role. Sometimes no genetic alteration is identified as a potential cause of disease after analysis of all known defects; thus, performing a wide-ranging analysis may allow the identification of possible causal but localized alterations in genes/chromosomes not definitively associated with suspected disease. Consequently, these alterations are again classified with an ambiguous role. Therefore, performing a complete genetic analysis is not always the answer to increase genetic yield. From our point of view, the best approach is to analyze known genes/chromosomes and perform an exhaustive interpretation before clinical translation. The analysis of unknown areas of the genome is essential to improve the genetic knowledge of CD, but it should be done in the field of research, not in clinical diagnosis.
Nowadays, CHDs require rapid diagnosis and treatment to improve their prognosis. This diagnosis can be prenatal, at the time of birth, or during the first years of childhood depending on clinical suspicions/diagnosis. More precise and earlier diagnosis allows adoption of immediate and appropriate therapeutic measures for each case, factors that increase the percentage of survival and quality of life. An estimated 65% of cases are diagnosed prenatally and 20% during the first week or month of life. Around a third of patients with CHD undergo at least one surgery during the first year of life; the results of these interventions are improving, which allows many patients to lead a relatively normal life and reach adulthood
[6].
2. Epidemiology
CHDs are the most common form of congenital malformations (approximately one third of the total) as well as a leading cause of morbidity and mortality during childhood. CHD occurs in 0.5–0.8% of live births but this value increases among abortions and premature infants. Specifically, the prevalence ranges from 8–10 per 1000 live births before the first year of life and 12 per 1000 before the age of 16, with a higher prevalence in industrialized countries
[7]. There is a higher incidence of these disorders in males than in females, as occurs with infant mortality. Likewise, differences in incidence between races have been reported, for example a higher male incidence is observed born to Asian mothers. In the Spanish population, the total incidence of CHD is 13 per 1000 live births, which is higher than the estimated worldwide incidence but similar to that of other countries such as Switzerland or Malta
[8].
New diagnostic techniques allow the identification of alterations during the first days/weeks of life. This early diagnosis combined with advances in surgical procedures for CHD repair
[9][10] have allowed the survival rate of patients with malformations of this type to reach almost 90%
[11]. This higher survival has led to a significant increase in the number of adults with CHD, as well as an increase in the average life expectancy when the appropriate measures are adopted and with earlier diagnosis. Despite these advances, not all regions of the world have the means to implement these diagnostic techniques and treatments, which is why CHD is one of the leading causes of mortality in children under one year of age in underdeveloped countries. Women have a greater survival rate than men after a similar surgical intervention, as well as a lower rate of incidents and adverse effects in adults with corrected CHD
[12].
3. Genetic Basis
Firstly, is important to state that genetic diagnosis is profoundly dependent on accurate clinical diagnosis. The genetic basis of CHD is currently divided into two groups: syndromic CHD and non-syndromic (or also isolated/sporadic) CHD
[13]. Nowadays, most cases of CHD are diagnosed as non-syndromic, sporadic defects. Thus, neither a family history nor a clear Mendelian inheritance may be recognized. The genetic etiology of CHD is estimated in almost 30% of diagnosed cases, some of them associated with other syndromes.
Syndromic CHD is defined as CHD with other congenital anomalies and/or dysmorphic features. The alterations responsible for syndromic CHD can be aneuploidies, large deletions or duplications of DNA (>1000 nucleotides) called copy number variants (CNVs), or single nucleotide variants (rare monogenic pathogenic variants, rare oligogenic deleterious variants, and common variants)
[14]. It is important to note that a single genetic defect can cause different CHDs and the same malformation can be caused by alterations in different genes. Both situations make it difficult to identify the specific origin of the malformation and complicate genetic counseling regarding transmission to descendants. In isolated CHD, pathogenic variants encode the transcription factors, signaling molecules, structural proteins and epigenetic modifiers that are essential for normal cardiac development
[13][15].
4. Cardiac Malformations
More than fifty different types of cardiac malformations have been described; among these, atrial septal defect (ASD) is the most frequent, followed by ventricular septal defect (VSD) and persistent ductus arteriosus (PDA). Sometimes the same patient can manifest more than one CHD and this accumulation of malformations is usually associated with a worse prognosis. Clinically, CHD presents symptoms of different severity, many of which are mild forms that remit in the first months of life and do not impact adult life. This is the case in foramen ovale, an interatrial communications disorder that has a high incidence despite its low severity.
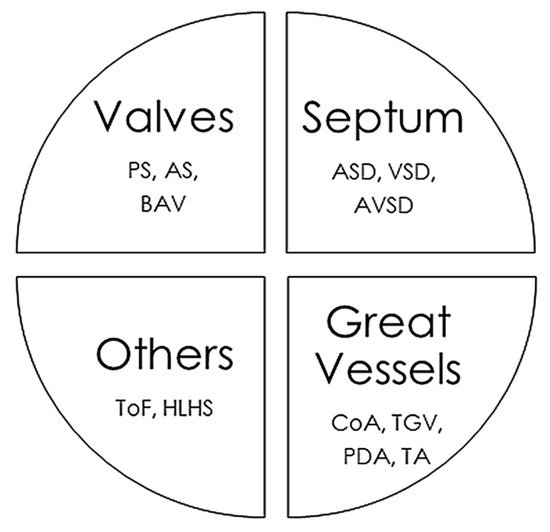
The CHD classifications vary widely depending on the different factors considered, such as structural alterations, severity, frequency in the population, and whether or not they cause cyanosis. In this review, we have classified CHDs taking into account the associated structural alterations. Some pathologies are associated with multiple defects, so their classification is somewhat more complex and they could be placed in several groups simultaneously (Figure 1).
Figure 1. Main congenital heart defects and anatomic alterations. AS, Aortic valvular stenosis; ASD, Atrial septal defect; AVSD, Atrioventricular septal defect; BAV, Bicuspid aortic valve; CoA, Coarctation of the aorta; HLHS, Hypoplastic left heart syndrome, PDA, Persistent ductus arteriosus; PS, Pulmonary stenosis; ToF, Tetralogy of Fallot; TGV, Transposition of great vessels; TA, Common Artery Trunk or Truncus arteriosus; VSD, Ventricular septal defect.
5. Syndromes with Congenital Cardiac Alteration
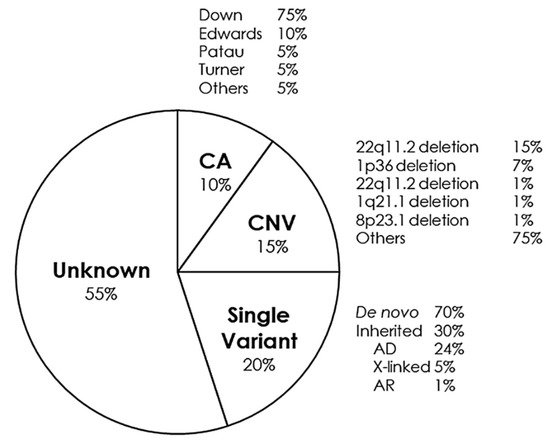
CHDs can occur in isolation, but approximately a quarter are associated with syndromes of genetic origin that lead to other extracardiac malformations. CHD can be found in 35–50% of children born with trisomy 21 (Down syndrome), in 60–80% of children born with trisomy 13 and 18 (Patau and Edwards syndrome, respectively) and in 33% of girls with monosomy X (Turner syndrome)
[16]. Some of the main syndromes that occur with CHD are detailed below (Figure 2).
Figure 2. Genetic distribution of congenital heart defects. AD, Autosomal Dominant; AR, Autosomal Recessive; CA, Chromosomic Alterations; CNV, Copy Number Variants.
6. Conclusions
CHDs are defects that occur during embryonic development of the heart. The etiology is multifactorial but genetic alterations are the main cause. Advances in clinical diagnosis and treatment have allowed a high percentage of patients to survive and reach adulthood with a good quality of life. Use of novel genetic technologies has made it possible to identify a multitude of genetic defects responsible for this type of malformation. This has increased the number of cases with a genetic diagnosis in the last ten years and has allowed early identification of cases, as well as offering adequate genetic counseling for affected families who wish to have offspring. Genetic diagnosis is heavily dependent on accurate clinical diagnosis. An early conclusive diagnosis as well as the adoption of personalized therapeutic measures is crucial in the favorable evolution of the newborn.
This entry is adapted from the peer-reviewed paper 10.3390/jpm11060562