 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Fengjuan Fan | + 2437 word(s) | 2437 | 2021-05-18 11:42:25 | | | |
| 2 | Vicky Zhou | + 1 word(s) | 2438 | 2021-05-20 09:54:19 | | |
Video Upload Options
Multiple myeloma (MM) is an incurable hematologic malignancy characterized by the clonal expansion of malignant plasma cells within the bone marrow. Activator Protein-1 (AP-1) transcription factors (TFs), comprised of the JUN, FOS, ATF and MAF multigene families, are implicated in a plethora of physiologic processes and tumorigenesis including plasma cell differentiation and MM pathogenesis. Depending on the genetic background, the tumor stage, and cues of the tumor microenvironment, specific dimeric AP-1 complexes are formed. For example, AP-1 complexes containing Fra-1, Fra-2 and B-ATF play central roles in the transcriptional control of B cell development and plasma cell differentiation, while dysregulation of AP-1 family members c-Maf, c-Jun, and JunB is associated with MM cell proliferation, survival, drug resistance, bone marrow angiogenesis, and bone disease. The present review article summarizes our up-to-date knowledge on the role of AP-1 family members in plasma cell differentiation and MM pathophysiology. Moreover, it discusses novel, rationally derived approaches to therapeutically target AP-1 TFs, including protein-protein and protein-DNA binding inhibitors, epigenetic modifiers and natural products.
1. Introduction
2. AP-1 in Plasma Cell Biology
AP-1 TFs play a critical role in PC formation and function. Specific functions of selected AP-1 TF family members during PC differentiation will be discussed below (Table 1 and Figure 1). For details, please refer to the original article (10.3390/cancers13102326) [23].
3. AP-1 in Multiple Myeloma
Besides acting as critical regulators in PC differentiation, AP-1 TFs are emerging as “master regulators” of aberrant gene expression programs in MM. Below we will discuss functions of AP-1 TFs that have specifically been associated with MM pathogenesis during recent years, c-Maf and MafB, c-Jun, JunB, in particular. Whether Fra-1, Fra-2, B-ATF and other AP-1 family members are deregulated in MM cells is currently unknown and subject of our own and others’ ongoing research efforts (Table 1 and Figure 1). For details, please refer to the original article (10.3390/cancers13102326) [23].
Table 1. Function of AP-1 in plasma cell biology and multiple myeloma pathophysiology.
| AP-1 Member | Activity | Mechanism | References |
|---|---|---|---|
| Plasma cell biology | |||
| Fra-1 | Suppresses B cell differentiation into PCs and decreases Ig production | Inhibition of Prdm1/Blimp-1 expression by preventing binding of c-Fos to the promoter | [24][25][26] |
| Fra-2 | Enhances B cell proliferation and differentiation at multiple stages |
Transcriptional induction of FOXO-1 and IRF-4 expression, and their downstream targets Ikaros, IL7Ra, Rag1/2 and Aiolos | [27] |
| B-ATF | Essential for GC formation and effective CSR |
Downstream of FOXO-1, modulating the expression of Aicda/AID and GLTs from the Ig locus of B cells in the GC | [28][29] |
| Regulates B cell activation and GC response |
Binding of B-ATF containing AP-1 complexes and IRF-4 to the AICE motif of target genes | [30][31] | |
| Multiple myeloma | |||
| c-Maf MafB |
Overexpressed in MM | Chromosomal translocation t(14;16), t(14;20) MMSET/MEK/ERK/AP-1 signaling sequelae |
[11][18][32] |
| Promote MM cell proliferation, migration and invasion, survival, adhesion and pathological interactions with BMSC |
Regulation of cyclin D2, ARK5, DEPTOR, and integrin β7 expression | [33][34][35] | |
| Confer resistance to PIs bortezomib and carfilzomib | Abrogation of GSK3β-mediated proteasomal degradation of c-Maf and MafB |
[36][37] | |
| c-Jun | Lower expression in primary MM cells compared to normal PCs | Unknown | [38] |
| Upregulated in MM cells by adaphostin or bortezomib Inhibits proliferation and induces apoptosis |
Caspase-mediated c-Abl cleavage Upregulation of EGR-1 Upregulation of p53 |
[39][40][41][42] | |
| JunB | BMSC- and IL-6- triggered upregulation in MM cells | MEK/MAPK- and NFκB- dependent | [43] |
| Promotes MM cell proliferation | Cell cycle regulation | ||
| Protects MM cells against dexamethasone- and bortezomib- induced cell death |
Inhibition of apoptotic pathways | ||
| Promotes MM BM angiogenesis | Transcriptional regulation of angiogenic factors VEGF, VEGFB and IGF1 | [44] | |
| Bone metabolism | |||
| c-Fos | Regulates OC differentiation (Block in OC differentiation in mice lacking c-Fos) |
Induced by RANKL and M-CSF Transcriptional regulation of Fra-1 and NFATc1 |
[45][46][47][48] |
| Fra-1 | Regulates OB activity and bone matrix formation (Mice overexpressing Fra-1 develop osteosclerosis) |
Regulation of bone matrix component production by OBs (osteocalcin, collagen1α2, and matrix Gla protein) | [49][50] |
| Fra-2 | Regulates OB differentiation (Fra-2-overexpressing mice are osteosclerotic) |
Transcriptional regulation of osteocalcin and collagen1α2 | [51] |
| Controls OC survival and size (Increased size and numbers of OCs in Fra-2-deficient mice) |
Transcriptional induction of LIF via Fra-2: c-Jun heterodimers Modulation of LIF/LIF-receptor/PHD2/HIF1α signaling sequelae |
[52] | |
| JunB | Regulates OB proliferation and differentiation (Mice lacking JunB are osteopenic) |
Cyclin D1 and cyclin A expression, and collagen1α2, osteocalcin and bone sialoprotein production |
[53] |
| Regulates OC proliferation and differentiation |
Dimerization partner of c-Fos (?) | ||
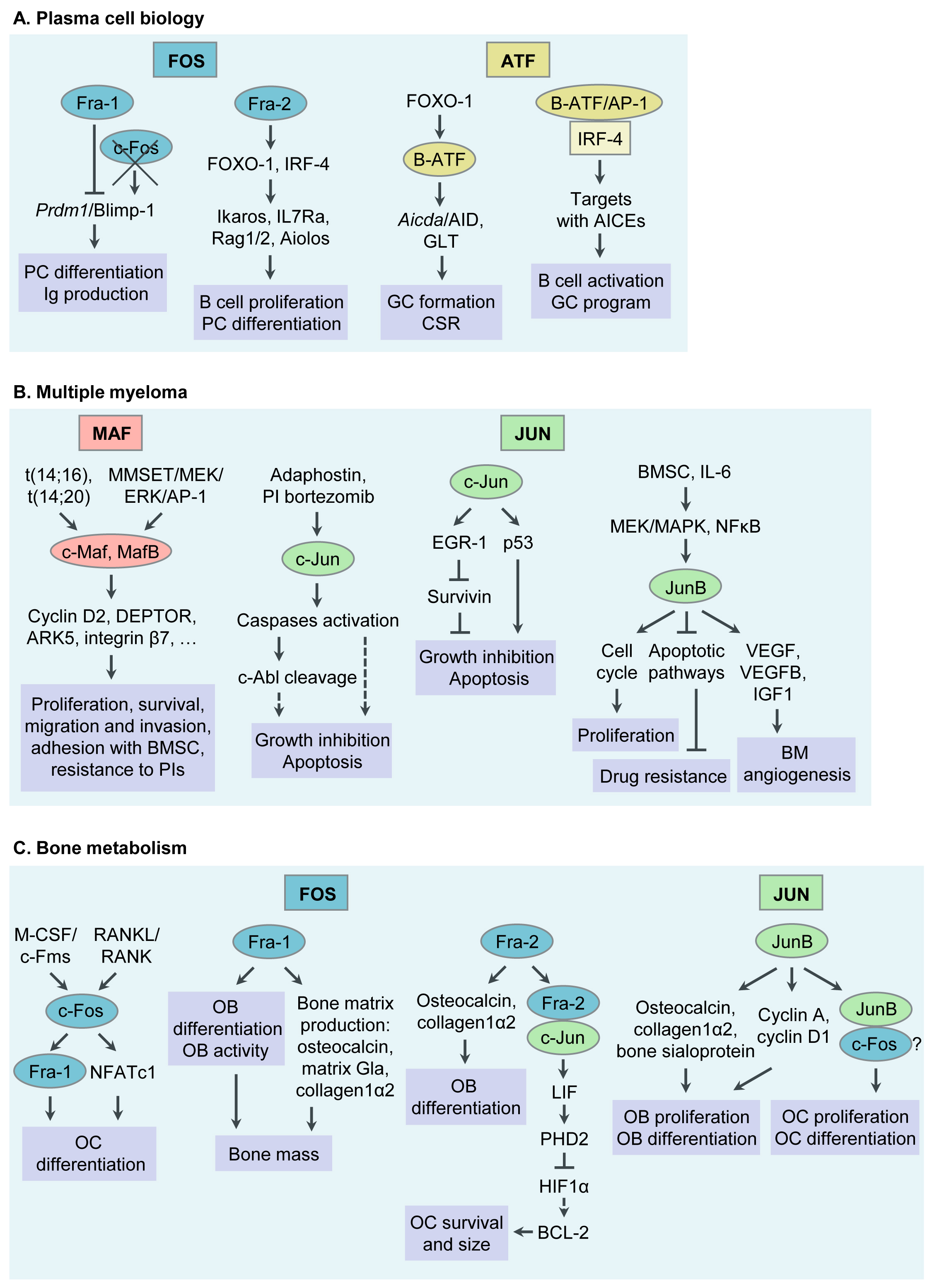
Figure 1. Functions of Activator Protein-1 (AP-1)/JUN, FOS, ATF and MAF transcription factor (TF) subfamily members in plasma cell (PC) biology, multiple myeloma (MM) pathophysiology, bone metabolism and MM associated bone disease. (A) Functions of AP-1 TFs in PC biology. (B) Functions of AP-1 TFs in MM pathogenesis. (C) Functions of AP-1 TFs in bone metabolism and MM associated bone disease. Ig, immunoglobulin; GC, germinal center; CSR, class switch recombination; AID, activation- induced cytidine deaminase; GLT, germline transcript; AICEs, AP-1-IRF composite elements; BM, bone marrow; BMSC, bone marrow stromal cell; PI, proteasome inhibitor; OC, osteoclast; RANKL, receptor activator of NFκB ligand; M-CSF, macrophage colony stimulating factor; NFAT, nuclear factor of activated T cells; LIF, leukaemia inhibitory factor; OB, osteoblast.
4. Targeting AP-1 TFs for MM Therapy
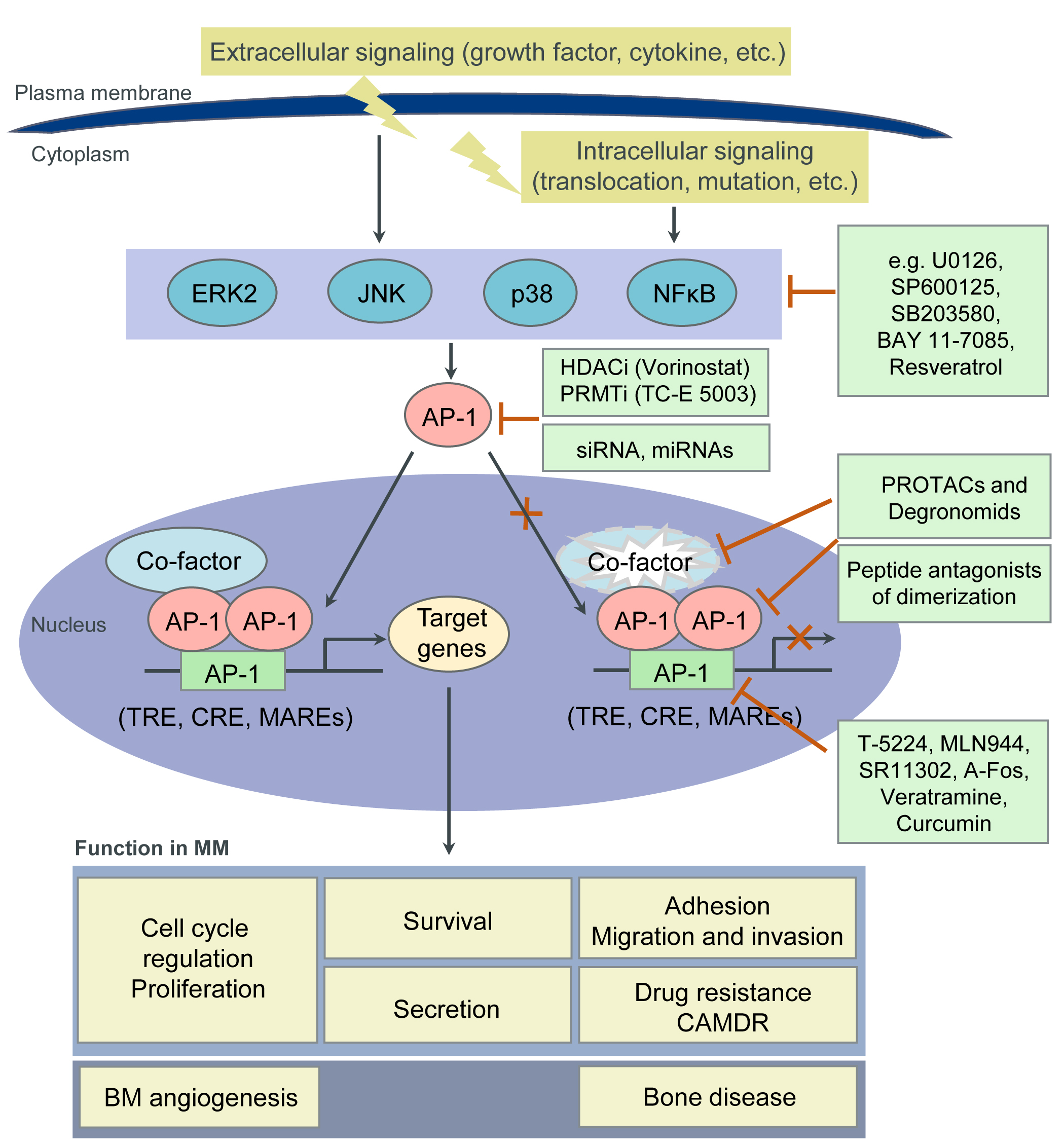
| Strategies | Inhibitors | Targets | References |
|---|---|---|---|
| Inhibition of protein-protein interactions |
Peptidic inhibitors of c-Maf dimerization |
Leucine zipper motif of c-Maf | [61] |
| Peptide antagonists of c-Jun dimerization |
Leucine zipper motif of c-Jun | [62][63][64][65] | |
| Peptide antagonists of c-Jun: c-Fos dimerization |
Leucine zipper motif of c-Jun or c-Fos | [66][67] | |
| Leucine zipper peptide (Superzipper) |
Leucine zipper dimerization domains of both c-Jun and c-Fos |
[68] | |
| Inhibition of protein- DNA binding |
T-5224 | bZIP domain of c-Fos/AP-1 -DNA complex |
[69][70] |
| MLN944 (XR5944) | TRE | [71] | |
| SR11302 | TRE | [72][73] | |
| Dominant negative peptide A-Fos | bZIP domain of c-Jun | [74] | |
| Regulation of epigenetic events | Valproic acid (VPA) Vorinostat (SAHA) Trichostatin A (TSA) LBH589 |
HDAC (Transcriptional suppression of c-Jun and Fra-1 expression) |
[75] |
| TC-E 5003 (TC-E) | PRMT (Suppression of c-Jun expression and nuclear translocation) |
[76] | |
| Natural products | Curcumin | Suppression of c-Fos and c-Jun expression and their binding to DNA |
[77] |
| Resveratrol | Suppression of c-Fos and c-Jun expression and AP-1 activity |
[78] | |
| Veratramine | TRE | [79] |
5. Conclusions
References
- Eferl, R.; Wagner, E.F. AP-1: A double-edged sword in tumorigenesis. Nat. Rev. Cancer 2003, 3, 859–868.
- Curran, T.; Peters, G.; Van Beveren, C.; Teich, N.M.; Verma, I.M. FBJ murine osteosarcoma virus: Identification and molecular cloning of biologically active proviral DNA. J. Virol. 1982, 44, 674–682.
- Maki, Y.; Bos, T.J.; Davis, C.; Starbuck, M.; Vogt, P.K. Avian sarcoma virus 17 carries the jun oncogene. Proc. Natl. Acad. Sci. USA 1987, 84, 2848–2852.
- Bohmann, D.; Bos, T.; Admon, A.; Nishimura, T.; Vogt, P.; Tjian, R. Human proto-oncogene c-jun encodes a DNA binding protein with structural and functional properties of transcription factor AP-1. Science 1987, 238, 1386–1392.
- Lee, W.; Mitchell, P.; Tjian, R. Purified transcription factor AP-1 interacts with TPA-inducible enhancer elements. Cell 1987, 49, 741–752.
- Atsaves, V.; Leventaki, V.; Rassidakis, G.Z.; Claret, F.X. AP-1 Transcription Factors as Regulators of Immune Responses in Cancer. Cancers 2019, 11, 1037.
- Shaulian, E.; Karin, M. AP-1 as a regulator of cell life and death. Nat. Cell Biol. 2002, 4, E131–E136.
- Jochum, W.; Passegué, E.; Wagner, E.F. AP-1 in mouse development and tumorigenesis. Oncogene 2001, 20, 2401–2412.
- Rajkumar, S.V. Multiple myeloma: 2020 update on diagnosis, risk-stratification and management. Am. J. Hematol. 2020, 95, 548–567.
- Bianchi, G.; Munshi, N.C. Pathogenesis beyond the cancer clone(s) in multiple myeloma. Blood 2015, 125, 3049–3058.
- Manier, S.; Salem, K.Z.; Park, J.; Landau, D.A.; Getz, G.; Ghobrial, I.M. Genomic complexity of multiple myeloma and its clinical implications. Nat. Rev. Clin. Oncol. 2017, 14, 100–113.
- Morgan, G.J.; Walker, B.A.; Davies, F.E. The genetic architecture of multiple myeloma. Nat. Rev. Cancer 2012, 12, 335–348.
- Gasparetto, C.; Lentzsch, S.; Schiller, G.; Callander, N.; Tuchman, S.; Chen, C.; White, D.; Kotb, R.; Sutherland, H.; Sebag, M.; et al. Selinexor, daratumumab, and dexamethasone in patients with relapsed or refractory multiple myeloma. eJHaem 2021, 2, 56–65.
- Markham, A. Belantamab Mafodotin: First Approval. Drugs 2020, 80, 1607–1613.
- Kuehl, W.M.; Bergsagel, P.L. Multiple myeloma: Evolving genetic events and host interactions. Nat. Rev. Cancer 2002, 2, 175–187.
- Shapiro-Shelef, M.; Calame, K. Regulation of plasma-cell development. Nat. Rev. Immunol. 2005, 5, 230–242.
- Li, S.; Vallet, S.; Sacco, A.; Roccaro, A.; Lentzsch, S.; Podar, K. Targeting transcription factors in multiple myeloma: Evolving therapeutic strategies. Expert Opin. Investig. Drugs 2019, 28, 445–462.
- Pawlyn, C.; Morgan, G.J. Evolutionary biology of high-risk multiple myeloma. Nat. Rev. Cancer 2017, 17, 543–556.
- Bradner, J.E.; Hnisz, D.; Young, R.A. Transcriptional Addiction in Cancer. Cell 2017, 168, 629–643.
- Shaffer, A.L.; Emre, N.C.T.; Lamy, L.; Ngo, V.N.; Wright, G.; Xiao, W.; Powell, J.; Dave, S.; Yu, X.; Zhao, H.; et al. IRF4 addiction in multiple myeloma. Nature 2008, 454, 226–231.
- Holien, T.; Våtsveen, T.K.; Hella, H.; Waage, A.; Sundan, A. Addiction to c-MYC in multiple myeloma. Blood 2012, 120, 2450–2453.
- Jovanović, K.K.; Roche-Lestienne, C.; Ghobrial, I.M.; Facon, T.; Quesnel, B.; Manier, S. Targeting MYC in multiple myeloma. Leukemia 2018, 32, 1295–1306.
- Fan, F.; Podar, K. The Role of AP-1 Transcription Factors in Plasma Cell Biology and Multiple Myeloma Pathophysiology. Cancers 2021, 13, 2326. https://doi.org/10.3390/cancers13102326
- Ohkubo, Y.; Arima, M.; Arguni, E.; Okada, S.; Yamashita, K.; Asari, S.; Obata, S.; Sakamoto, A.; Hatano, M.; Wang, J.O.; et al. A Role for c- fos/Activator Protein 1 in B Lymphocyte Terminal Differentiation. J. Immunol. 2005, 174, 7703–7710.
- Grötsch, B.; Brachs, S.; Lang, C.; Luther, J.; Derer, A.; Schlötzer-Schrehardt, U.; Bozec, A.; Fillatreau, S.; Berberich, I.; Hobeika, E.; et al. The AP-1 transcription factor Fra1 inhibits follicular B cell differentiation into plasma cells. J. Exp. Med. 2014, 211, 2199–2212.
- Wagner, E.F. Bone development and inflammatory disease is regulated by AP-1 (Fos/Jun). Ann. Rheum. Dis. 2010, 69, i86–i88.
- Ubieta, K.; Garcia, M.; Grötsch, B.; Uebe, S.; Weber, G.F.; Stein, M.; Ekici, A.; Schett, G.; Mielenz, D.; Bozec, A. Fra-2 regulates B cell development by enhancing IRF4 and Foxo1 transcription. J. Exp. Med. 2017, 214, 2059–2071.
- Ise, W.; Kohyama, M.; Schraml, B.U.; Zhang, T.; Schwer, B.; Basu, U.; Alt, F.W.; Tang, J.; Oltz, E.M.; Murphy, T.L.; et al. The transcription factor BATF controls the global regulators of class-switch recombination in both B cells and T cells. Nat. Immunol. 2011, 12, 536–543.
- Dominguez-Sola, D.; Kung, J.; Holmes, A.B.; Wells, V.A.; Mo, T.; Basso, K.; Dalla-Favera, R. The FOXO1 Transcription Factor Instructs the Germinal Center Dark Zone Program. Immunity 2015, 43, 1064–1074.
- Ochiai, K.; Maienschein-Cline, M.; Simonetti, G.; Chen, J.; Rosenthal, R.; Brink, R.; Chong, A.S.; Klein, U.; Dinner, A.R.; Singh, H.; et al. Transcriptional Regulation of Germinal Center B and Plasma Cell Fates by Dynamical Control of IRF4. Immunity 2013, 38, 918–929.
- Agnarelli, A.; Chevassut, T.; Mancini, E.J. IRF4 in multiple myeloma—Biology, disease and therapeutic target. Leuk. Res. 2018, 72, 52–58.
- Annunziata, C.M.; Hernandez, L.; Davis, R.E.; Zingone, A.; Lamy, L.; Lam, L.T.; Hurt, E.M.; Shaffer, A.L.; Kuehl, W.M.; Staudt, L.M. A mechanistic rationale for MEK inhibitor therapy in myeloma based on blockade of MAF oncogene expression. Blood 2011, 117, 2396–2404.
- Hurt, E.M.; Wiestner, A.; Rosenwald, A.; Shaffer, A.; Campo, E.; Grogan, T.; Bergsagel, P.L.; Kuehl, W.M.; Staudt, L.M. Overexpression of c-maf is a frequent oncogenic event in multiple myeloma that promotes proliferation and pathological interactions with bone marrow stroma. Cancer Cell 2004, 5, 191–199.
- Suzuki, A.; Iida, S.; Kato-Uranishi, M.; Tajima, E.; Zhan, F.; Hanamura, I.; Huang, Y.; Ogura, T.; Takahashi, S.; Ueda, R.; et al. ARK5 is transcriptionally regulated by the Large-MAF family and mediates IGF-1-induced cell invasion in multiple myeloma: ARK5 as a new molecular determinant of malignant multiple myeloma. Oncogene 2005, 24, 6936–6944.
- Peterson, T.R.; Laplante, M.; Thoreen, C.C.; Sancak, Y.; Kang, S.A.; Kuehl, W.M.; Gray, N.S.; Sabatini, D.M. DEPTOR Is an mTOR Inhibitor Frequently Overexpressed in Multiple Myeloma Cells and Required for Their Survival. Cell 2009, 137, 873–886.
- Qiang, Y.; Ye, S.; Chen, Y.; Buros, A.F.; Edmonson, R.; van Rhee, F.; Barlogie, B.; Epstein, J.; Morgan, G.J.; Davies, F.E. MAF protein mediates innate resistance to proteasome inhibition therapy in multiple myeloma. Blood 2016, 128, 2919–2930.
- Qiang, Y.-W.; Ye, S.; Huang, Y.; Chen, Y.; Van Rhee, F.; Epstein, J.; Walker, B.A.; Morgan, G.J.; Davies, F.E. MAFb protein confers intrinsic resistance to proteasome inhibitors in multiple myeloma. BMC Cancer 2018, 18, 724.
- Miannay, B.; Minvielle, S.; Roux, O.; Drouin, P.; Avet-Loiseau, H.; Guérin-Charbonnel, C.; Gouraud, W.; Attal, M.; Facon, T.; Munshi, N.C.; et al. Logic programming reveals alteration of key transcription factors in multiple myeloma. Sci. Rep. 2017, 7, 9257.
- Podar, K.; Raab, M.S.; Tonon, G.; Sattler, M.; Barilà, D.; Zhang, J.; Tai, Y.-T.; Yasui, H.; Raje, N.; DePinho, R.A.; et al. Up-Regulation of c-Jun Inhibits Proliferation and Induces Apoptosis via Caspase-Triggered c-Abl Cleavage in Human Multiple Myeloma. Cancer Res. 2007, 67, 1680–1688.
- Fan, F.; Tonon, G.; Bashari, M.H.; Vallet, S.; Antonini, E.; Goldschmidt, H.; Schulze-Bergkamen, H.; Opferman, J.T.; Sattler, M.; Anderson, K.C.; et al. Targeting Mcl-1 for multiple myeloma (MM) therapy: Drug-induced generation of Mcl-1 fragment Mcl-1128-350 triggers MM cell death via c-Jun upregulation. Cancer Lett. 2014, 343, 286–294.
- Chen, L.; Wang, S.; Zhou, Y.; Wu, X.; Entin, I.; Epstein, J.; Yaccoby, S.; Xiong, W.; Barlogie, B.; Shaughnessy, J.D.; et al. Identification of early growth response protein 1 (EGR-1) as a novel target for JUN-induced apoptosis in multiple myeloma. Blood 2010, 115, 61–70.
- Saha, M.N.; Jiang, H.; Yang, Y.; Zhu, X.; Wang, X.; Schimmer, A.D.; Qiu, L.; Chang, H. Targeting p53 via JNK Pathway: A Novel Role of RITA for Apoptotic Signaling in Multiple Myeloma. PLoS ONE 2012, 7, e30215.
- Fan, F.; Bashari, M.H.; Morelli, E.; Tonon, G.; Malvestiti, S.; Vallet, S.; Jarahian, M.; Seckinger, A.; Hose, D.; Bakiri, L.; et al. The AP-1 transcription factor JunB is essential for multiple myeloma cell proliferation and drug resistance in the bone marrow microenvironment. Leukemia 2017, 31, 1570–1581.
- Fan, F.; Malvestiti, S.; Vallet, S.; Lind, J.; Garcia-Manteiga, J.M.; Morelli, E.; Jiang, Q.; Seckinger, A.; Hose, D.; Goldschmidt, H.; et al. JunB is a key regulator of multiple myeloma bone marrow angiogenesis. Leukemia 2021. https://doi.org/10.1038/s41375-021-01271-9; https://rdcu.be/ckOLI
- Grigoriadis, A.; Wang, Z.; Cecchini, M.; Hofstetter, W.; Felix, R.; Fleisch, H.; Wagner, E. c-Fos: A key regulator of osteoclast-macrophage lineage determination and bone remodeling. Science 1994, 266, 443–448.
- Matsuo, K.; Owens, J.M.; Tonko, M.; Elliott, C.; Chambers, T.J.; Wagner, E.F. Fosl1 is a transcriptional target of c-Fos during osteoclast differentiation. Nat. Genet. 2000, 24, 184–187.
- Takayanagi, H.; Kim, S.; Matsuo, K.; Suzuki, H.; Suzuki, T.; Sato, K.; Yokochi, T.; Oda, H.; Nakamura, K.; Ida, N.; et al. RANKL maintains bone homeostasis through c-Fos-dependent induction of interferon-β. Nature 2002, 416, 744–749.
- Takayanagi, H.; Kim, S.; Koga, T.; Nishina, H.; Isshiki, M.; Yoshida, H.; Saiura, A.; Isobe, M.; Yokochi, T.; Inoue, J.; et al. Induction and Activation of the Transcription Factor NFATc1 (NFAT2) Integrate RANKL Signaling in Terminal Differentiation of Osteoclasts. Dev. Cell 2002, 3, 889–901.
- Jochum, W.; David, J.-P.; Elliott, C.; Wutz, A.; Plenk, H.; Matsuo, K.; Wagner, E.F. Increased bone formation and osteosclerosis in mice overexpressing the transcription factor Fra-1. Nat. Med. 2000, 6, 980–984.
- Eferl, R.; Hoebertz, A.; Schilling, A.F.; Rath, M.; Karreth, F.; Kenner, L.; Amling, M.; Wagner, E.F. The Fos-related antigen Fra-1 is an activator of bone matrix formation. EMBO J. 2004, 23, 2789–2799.
- Bozec, A.; Bakiri, L.; Jimenez, M.; Schinke, T.; Amling, M.; Wagner, E.F. Fra-2/AP-1 controls bone formation by regulating osteoblast differentiation and collagen production. J. Cell Biol. 2010, 190, 1093–1106.
- Bozec, A.; Bakiri, L.; Hoebertz, A.; Eferl, R.; Schilling, A.F.; Komnenovic, V.; Scheuch, H.; Priemel, M.; Stewart, C.L.; Amling, M.; et al. Osteoclast size is controlled by Fra-2 through LIF/LIF-receptor signalling and hypoxia. Nature 2008, 454, 221–225.
- Kenner, L.; Hoebertz, A.; Beil, F.T.; Keon, N.; Karreth, F.; Eferl, R.; Scheuch, H.; Szremska, A.; Amling, M.; Schorpp-Kistner, M.; et al. Mice lacking JunB are osteopenic due to cell-autonomous osteoblast and osteoclast defects. J. Cell Biol. 2004, 164, 613–623.
- Ye, N.; Ding, Y.; Wild, C.; Shen, Q.; Zhou, J. Small Molecule Inhibitors Targeting Activator Protein 1 (AP-1). J. Med. Chem. 2014, 57, 6930–6948.
- Chen, A.; Koehler, A.N. Transcription Factor Inhibition: Lessons Learned and Emerging Targets. Trends Mol. Med. 2020, 26, 508–518.
- Dang, C.V.; Reddy, E.P.; Shokat, K.M.; Soucek, L. Drugging the “undruggable” cancer targets. Nat. Rev. Cancer 2017, 17, 502–508.
- Gonda, T.J.; Ramsay, R.G. Directly targeting transcriptional dysregulation in cancer. Nat. Rev. Cancer 2015, 15, 686–694.
- Bushweller, J.H. Targeting transcription factors in cancer—From undruggable to reality. Nat. Rev. Cancer 2019, 19, 611–624.
- Kim, E.; Ahuja, A.; Kim, M.-Y.; Cho, J.Y. DNA or Protein Methylation-Dependent Regulation of Activator Protein-1 Function. Cells 2021, 10, 461.
- Serafimova, I.M.; Pufall, M.A.; Krishnan, S.; Duda, K.; Cohen, M.S.; Maglathlin, R.L.; McFarland, J.M.; Miller, R.M.; Frödin, M.; Taunton, J. Reversible targeting of noncatalytic cysteines with chemically tuned electrophiles. Nat. Chem. Biol. 2012, 8, 471–476.
- Pellegrino, S.; Ronda, L.; Annoni, C.; Contini, A.; Erba, E.; Gelmi, M.L.; Piano, R.; Paredi, G.; Mozzarelli, A.; Bettati, S. Molecular insights into dimerization inhibition of c-Maf transcription factor. Biochim. Biophys. Acta Proteins Proteomics 2014, 1844, 2108–2115.
- Lathbridge, A.; Mason, J.M. Computational Competitive and Negative Design To Derive a Specific cJun Antagonist. Biochemistry 2018, 57, 6108–6118.
- Baxter, D.; Perry, S.R.; Hill, T.A.; Kok, W.M.; Zaccai, N.R.; Brady, R.L.; Fairlie, D.P.; Mason, J.M. Downsizing Proto-oncogene cFos to Short Helix-Constrained Peptides That Bind Jun. ACS Chem. Biol. 2017, 12, 2051–2061.
- Baxter, D.; Ullman, C.G.; Frigotto, L.; Mason, J.M. Exploiting Overlapping Advantages of In Vitro and In Cellulo Selection Systems to Isolate a Novel High-Affinity cJun Antagonist. ACS Chem. Biol. 2017, 12, 2579–2588.
- Pernelle, C.; Clerc, F.F.; Dureuil, C.; Bracco, L.; Tocque, B. An efficient screening assay for the rapid and precise determination of affinities between leucine zipper domains. Biochemistry 1993, 32, 11682–1168
- Worrall, J.A.R.; Mason, J.M. Thermodynamic analysis of Jun-Fos coiled coil peptide antagonists. FEBS J. 2011, 278, 663–672.
- Mason, J.M.; Schmitz, M.A.; Muller, K.M.; Arndt, K.M. Semirational design of Jun-Fos coiled coils with increased affinity: Universal implications for leucine zipper prediction and design. Proc. Natl. Acad. Sci. USA 2006, 103, 8989–8994.
- Bains, N.P.S.; Wilce, J.A.; Heuer, K.H.; Tunstall, M.; Mackey, J.P.; Bennett, M.R.; Weiss, A.S.; King, G.F. Zipping up transcription factors: Rational design of anti-Jun and anti-Fos peptides. Lett. Pept. Sci. 1997, 4, 67–77.
- Uchihashi, S.; Fukumoto, H.; Onoda, M.; Hayakawa, H.; Ikushiro, S.; Sakaki, T. Metabolism of the c-Fos/Activator Protein-1 Inhibitor T-5224 by Multiple Human UDP-Glucuronosyltransferase Isoforms. Drug Metab. Dispos. 2011, 39, 803–813.
- Izuta, S.; Ueki, M.; Ueno, M.; Nishina, K.; Shiozawa, S.; Maekawa, N. T-5224, a selective inhibitor of c-Fos/activator protein-1, attenuates lipopolysaccharide-induced liver injury in mice. Biotechnol. Lett. 2012, 34, 2175–2182.
- Dai, J.; Punchihewa, C.; Mistry, P.; Ooi, A.T.; Yang, D. Novel DNA bis-intercalation by MLN944, a potent clinical bisphenazine anticancer drug. J. Biol. Chem. 2004, 279, 46096–46103.
- Fanjul, A.; Dawson, M.I.; Hobbs, P.D.; Jong, L.; Cameron, J.F.; Harlev, E.; Graupner, G.; Lu, X.-P.; Pfahl, M. A new class of retinoids with selective inhibition of AP-1 inhibits proliferation. Nature 1994, 372, 107–111.
- Huang, C.; Ma, W.-Y.; Dawson, M.I.; Rincon, M.; Flavell, R.A.; Dong, Z. Blocking activator protein-1 activity, but not activating retinoic acid response element, is required for the antitumor promotion effect of retinoic acid. Proc. Natl. Acad. Sci. USA 1997, 94, 5826–5830.
- Olive, M.; Krylov, D.; Vinson, C.; Echlin, D.R.; Gardner, K.; Taparowsky, E. A Dominant Negative to Activation Protein-1 (AP1) That Abolishes DNA Binding and Inhibits Oncogenesis. J. Biol. Chem. 1997, 272, 18586–18594.
- He, W.; Wu, Y.; Tang, X.; Xia, Y.; He, G.; Min, Z.; Li, C.; Xiong, S.; Shi, Z.; Lu, Y.; et al. HDAC inhibitors suppress c-Jun/Fra-1-mediated proliferation through transcriptionally downregulating MKK7 and Raf1 in neuroblastoma cells. Oncotarget 2016, 7, 6727–6747.
- Kim, E.; Jang, J.; Park, J.G.; Kim, K.-H.; Yoon, K.; Yoo, B.C.; Cho, J.Y. Protein Arginine Methyltransferase 1 (PRMT1) Selective Inhibitor, TC-E 5003, Has Anti-Inflammatory Properties in TLR4 Signaling. Int. J. Mol. Sci. 2020, 21, 3058.
- Han, S.-S.; Keum, Y.-S.; Seo, H.-J.; Surh, Y.-J. Curcumin Suppresses Activation of NF-κB and AP-1 Induced by Phorbol Ester in Cultured Human Promyelocytic Leukemia Cells. J. Biochem. Mol. Biol. 2002, 35, 337–342.
- Boissy, P.; Andersen, T.L.; Abdallah, B.M.; Kassem, M.; Plesner, T.; Delaissé, J.-M. Resveratrol Inhibits Myeloma Cell Growth, Prevents Osteoclast Formation, and Promotes Osteoblast Differentiation. Cancer Res. 2005, 65, 9943–9952.
- Bai, F.; Liu, K.; Li, H.; Wang, J.; Zhu, J.; Hao, P.; Zhu, L.; Zhang, S.; Shan, L.; Ma, W.; et al. Veratramine modulates AP-1-dependent gene transcription by directly binding to programmable DNA. Nucleic Acids Res. 2018, 46, 546–557.


