Multiple myeloma (MM) is an incurable hematologic malignancy characterized by the clonal expansion of malignant plasma cells within the bone marrow. Activator Protein-1 (AP-1) transcription factors (TFs), comprised of the JUN, FOS, ATF and MAF multigene families, are implicated in a plethora of physiologic processes and tumorigenesis including plasma cell differentiation and MM pathogenesis. Depending on the genetic background, the tumor stage, and cues of the tumor microenvironment, specific dimeric AP-1 complexes are formed. For example, AP-1 complexes containing Fra-1, Fra-2 and B-ATF play central roles in the transcriptional control of B cell development and plasma cell differentiation, while dysregulation of AP-1 family members c-Maf, c-Jun, and JunB is associated with MM cell proliferation, survival, drug resistance, bone marrow angiogenesis, and bone disease. The present review article summarizes our up-to-date knowledge on the role of AP-1 family members in plasma cell differentiation and MM pathophysiology. Moreover, it discusses novel, rationally derived approaches to therapeutically target AP-1 TFs, including protein-protein and protein-DNA binding inhibitors, epigenetic modifiers and natural products.
- activator protein 1 (AP-1)
- transcription factor (TF)
- plasma cell (PC)
- multiple myeloma (MM)
- bone marrow (BM)
- microenvironment
1. Introduction
2. AP-1 in Plasma Cell Biology
AP-1 TFs play a critical role in PC formation and function. Specific functions of selected AP-1 TF family members during PC differentiation will be discussed below (Table 1 Table 1 and Figure 1 Figure 1). For details, please refer to the original article (10.3390/cancers13102326) [23].
3. AP-1 in Multiple Myeloma
Besides acting as critical regulators in PC differentiation, AP-1 TFs are emerging as “master regulators” of aberrant gene expression programs in MM. Below we will discuss functions of AP-1 TFs that have specifically been associated with MM pathogenesis during recent years, c-Maf and MafB, c-Jun, JunB, in particular. Whether Fra-1, Fra-2, B-ATF and other AP-1 family members are deregulated in MM cells is currently unknown and subject of our own and others’ ongoing research efforts (Table 1 and Figure 1). For details, please refer to the original article (10.3390/cancers13102326) [23].
Table 1.
Function of AP-1 in plasma cell biology and multiple myeloma pathophysiology.
| 66 | |||
| ] | |||
| [ | |||
| 67 | ] | ||
| Leucine zipper peptide (Superzipper) |
Leucine zipper dimerization domains of both c-Jun and c-Fos |
[68] | |
| Inhibition of protein- DNA binding |
T-5224 | bZIP domain of c-Fos/AP-1 -DNA complex |
[69][70] |
| MLN944 (XR5944) | TRE | [71] | |
| SR11302 | TRE | [72][73] | |
| Dominant negative peptide A-Fos | bZIP domain of c-Jun | [74] | |
| Regulation of epigenetic events | Valproic acid (VPA) Vorinostat (SAHA) Trichostatin A (TSA) LBH589 |
HDAC (Transcriptional suppression of c-Jun and Fra-1 expression) |
[75] |
| TC-E 5003 (TC-E) | PRMT (Suppression of c-Jun expression and nuclear translocation) |
[76] | |
| Natural products | Curcumin | Suppression of c-Fos and c-Jun expression and their binding to DNA |
[77] |
| Resveratrol | Suppression of c-Fos and c-Jun expression and AP-1 activity |
[78] | |
| Veratramine | TRE | [79] |
| AP-1 Member | Activity | Mechanism | References |
|---|
| Plasma cell biology | |||||
| Fra-1 | Suppresses B cell differentiation into PCs and decreases Ig production | Inhibition of | Prdm1 | /Blimp-1 expression by preventing binding of c-Fos to the promoter | [24][25][26] |
| Fra-2 | Enhances B cell proliferation and differentiation at multiple stages |
Transcriptional induction of FOXO-1 and IRF-4 expression, and their downstream targets Ikaros, IL7Ra, Rag1/2 and Aiolos | [27] | ||
| B-ATF | Essential for GC formation and effective CSR |
Downstream of FOXO-1, modulating the expression of | Aicda | /AID and GLTs from the Ig locus of B cells in the GC | [28][29] |
| Regulates B cell activation and GC response |
Binding of B-ATF containing AP-1 complexes and IRF-4 to the AICE motif of target genes | [30][31] | |||
| Multiple myeloma | |||||
| c-Maf MafB |
Overexpressed in MM | Chromosomal translocation t(14;16), t(14;20) MMSET/MEK/ERK/AP-1 signaling sequelae |
[11][18][32] | ||
| Promote MM cell proliferation, migration and invasion, survival, adhesion and pathological interactions with BMSC |
Regulation of cyclin D2, ARK5, DEPTOR, and integrin β7 expression | [33][34][35] | |||
| Confer resistance to PIs bortezomib and carfilzomib | Abrogation of GSK3β-mediated proteasomal degradation of c-Maf and MafB |
[36][37] | |||
| c-Jun | Lower expression in primary MM cells compared to normal PCs | Unknown | [38] | ||
| Upregulated in MM cells by adaphostin or bortezomib Inhibits proliferation and induces apoptosis |
Caspase-mediated c-Abl cleavage Upregulation of EGR-1 Upregulation of p53 |
[39][40][41][42] | |||
| JunB | BMSC- and IL-6- triggered upregulation in MM cells | MEK/MAPK- and NFκB- dependent | [43] | ||
| Promotes MM cell proliferation | Cell cycle regulation | ||||
| Protects MM cells against dexamethasone- and bortezomib- induced cell death |
Inhibition of apoptotic pathways | ||||
| Promotes MM BM angiogenesis | Transcriptional regulation of angiogenic factors VEGF, VEGFB and IGF1 | [44] | |||
| Bone metabolism | |||||
| c-Fos | Regulates OC differentiation (Block in OC differentiation in mice lacking c-Fos) |
Induced by RANKL and M-CSF Transcriptional regulation of Fra-1 and NFATc1 |
[45][46][47][48] | ||
| Fra-1 | Regulates OB activity and bone matrix formation (Mice overexpressing Fra-1 develop osteosclerosis) |
Regulation of bone matrix component production by OBs (osteocalcin, collagen1α2, and matrix Gla protein) | [49][50] | ||
| Fra-2 | Regulates OB differentiation (Fra-2-overexpressing mice are osteosclerotic) |
Transcriptional regulation of osteocalcin and collagen1α2 | [51] | ||
| Controls OC survival and size (Increased size and numbers of OCs in Fra-2-deficient mice) |
Transcriptional induction of LIF via Fra-2: c-Jun heterodimers Modulation of LIF/LIF-receptor/PHD2/HIF1α signaling sequelae |
[52] | |||
| JunB | Regulates OB proliferation and differentiation (Mice lacking JunB are osteopenic) |
Cyclin D1 and cyclin A expression, and collagen1α2, osteocalcin and bone sialoprotein production |
[53] | ||
| Regulates OC proliferation and differentiation |
Dimerization partner of c-Fos (?) | ||||
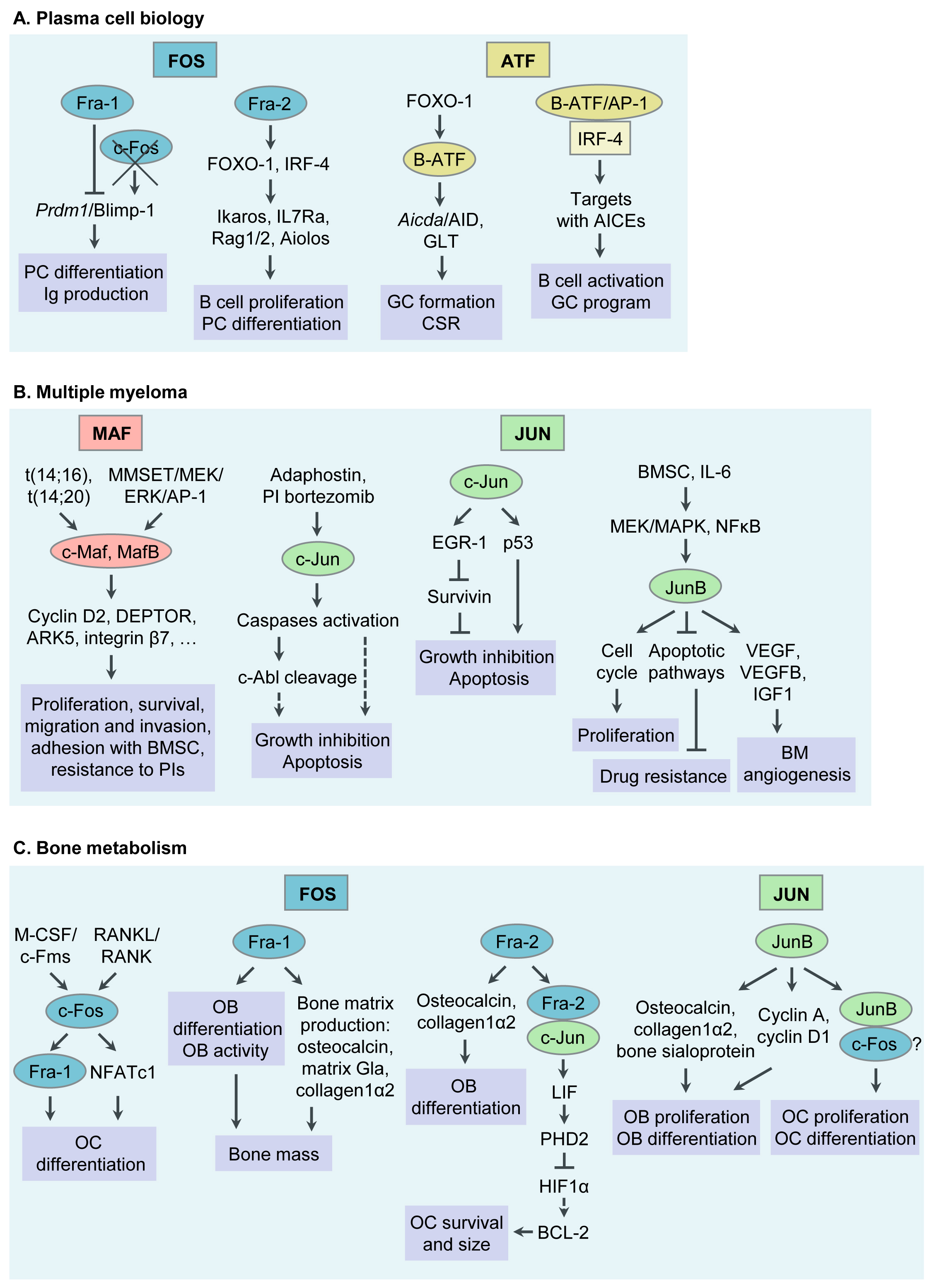
Figure 1. Functions of Activator Protein-1 (AP-1)/JUN, FOS, ATF and MAF transcription factor (TF) subfamily members in plasma cell (PC) biology, multiple myeloma (MM) pathophysiology, bone metabolism and MM associated bone disease. (A) Functions of AP-1 TFs in PC biology. (B) Functions of AP-1 TFs in MM pathogenesis. (C) Functions of AP-1 TFs in bone metabolism and MM associated bone disease. Ig, immunoglobulin; GC, germinal center; CSR, class switch recombination; AID, activation- induced cytidine deaminase; GLT, germline transcript; AICEs, AP-1-IRF composite elements; BM, bone marrow; BMSC, bone marrow stromal cell; PI, proteasome inhibitor; OC, osteoclast; RANKL, receptor activator of NFκB ligand; M-CSF, macrophage colony stimulating factor; NFAT, nuclear factor of activated T cells; LIF, leukaemia inhibitory factor; OB, osteoblast.
4. Targeting AP-1 TFs for MM Therapy
Accumulating evidence demonstrates a crucial role of deregulated AP-1 TFs in tumorigenesis in general, and MM in particular. AP-1 TFs therefore represent appealing therapeutic targets. However, TFs have been considered “undruggable” until recently due to their structural disorder (three-dimensional (3D) structure and architecture are very labile and dependent on TF interaction with functional proteins), their lack of tractable active sites (large protein-protein interfaces, lack of deep protein pockets) and their intracellular (often nuclear) localization. Nevertheless, with the progress of our understanding of the biochemical and biological properties of TFs, this paradigm does not hold true any longer. Indeed, members of the AP-1 family have emerged as worldwide actively pursued therapeutic targets, with a potentially high therapeutic index [17][54][55][56]. In MM, our own and other studies suggest therapeutic strategies that inhibit c-Maf or JunB and induce c-Jun activity. Besides inhibiting their expression (i.e., by siRNAs, miRNAs), novel approaches to target TFs in general, and AP-1 TFs in particular, include: (1) the disruption of either their interaction with functionally critical protein binding partners or; (2) their binding to the DNA (oligodeoxynucleotide decoys, pyrrole-imidazole polyamides or small molecules); (3) the modulation of their epigenetic binding through DNA methylation, histone methylation or modification; (4) the induction of proteasomal degradation of TFs by altering their ubiquitylation; as well as by utilizing PROteolysis-TArgeting Chimaeras (PROTACs) or Degronomids; (5) the inhibition of TF expression by modulating their regulators (i.e., MAPK- or NFκB-signaling molecules); (6) the use of reversible covalent drugs directed against non-conserved cysteines; and (7) the modulation of TF auto-inhibition. Moreover, disordered regions within TFs, which become structured upon interaction with binding partners (“coupled folding and binding”) may also represent attractive therapeutic targets. Indeed, these regions have a higher proportion of potential cavities and can more easily adjust to small molecules [56][57][58][59][60]. Below we discuss some potential approaches to target AP-1 TFs in MM (Figure 2 and Table 2). For details, please refer to the original article (10.3390/cancers13102326) [23].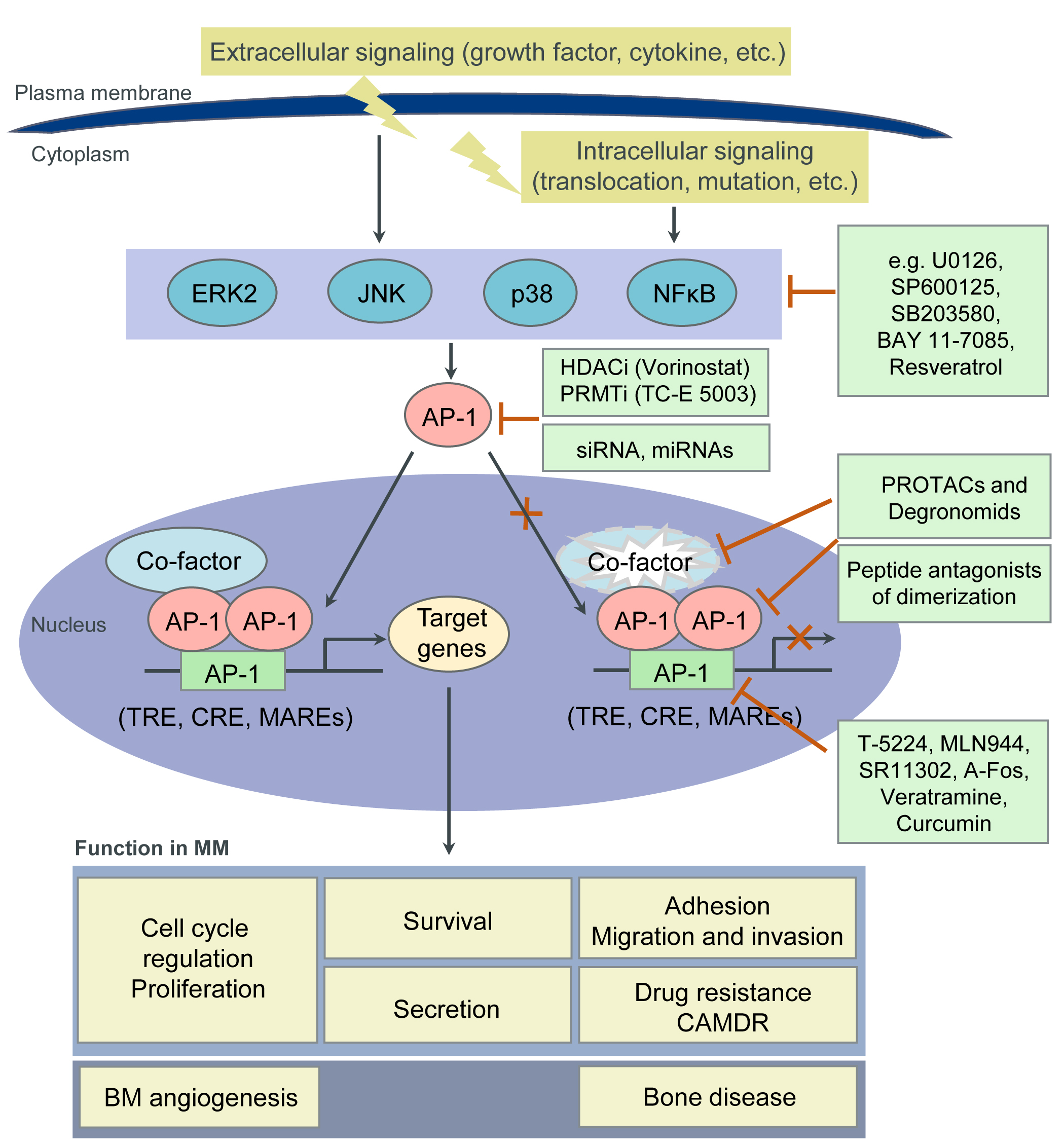
| Strategies | Inhibitors | Targets | References |
|---|
| Inhibition of protein-protein interactions |
Peptidic inhibitors of c-Maf dimerization |
Leucine zipper motif of c-Maf | [61] |
| Peptide antagonists of c-Jun dimerization |
Leucine zipper motif of c-Jun | [62][63][64][65] | |
| Peptide antagonists of c-Jun: c-Fos dimerization |
Leucine zipper motif of c-Jun or c-Fos | [ |
5. Conclusions
AP-1 TFs play essential roles in the transcriptional control of GC B cell development and PC differentiation. Dysregulation of AP-1 is an important mechanism in the oncogenic transformation and drug resistance of MM. Although recent discoveries are exciting, the therapeutic exploration of AP-1 TFs has just begun. Continuing basic and translational research on AP-1 TFs utilizing new technologies such as NMR-based screens, differential scanning fluorimetry (DSF), in silico 3D modelling, as well as Slim-seq will be fundamental to further advance our insights on the complex function of this TF family, facilitating the identification of valuable targets and the development of derived innovative therapies for MM to once more improve patient outcome.References
- Eferl, R.; Wagner, E.F. AP-1: A double-edged sword in tumorigenesis. Nat. Rev. Cancer 2003, 3, 859–868.
- Curran, T.; Peters, G.; Van Beveren, C.; Teich, N.M.; Verma, I.M. FBJ murine osteosarcoma virus: Identification and molecular cloning of biologically active proviral DNA. J. Virol. 1982, 44, 674–682.
- Maki, Y.; Bos, T.J.; Davis, C.; Starbuck, M.; Vogt, P.K. Avian sarcoma virus 17 carries the jun oncogene. Proc. Natl. Acad. Sci. USA 1987, 84, 2848–2852.
- Bohmann, D.; Bos, T.; Admon, A.; Nishimura, T.; Vogt, P.; Tjian, R. Human proto-oncogene c-jun encodes a DNA binding protein with structural and functional properties of transcription factor AP-1. Science 1987, 238, 1386–1392.
- Lee, W.; Mitchell, P.; Tjian, R. Purified transcription factor AP-1 interacts with TPA-inducible enhancer elements. Cell 1987, 49, 741–752.
- Atsaves, V.; Leventaki, V.; Rassidakis, G.Z.; Claret, F.X. AP-1 Transcription Factors as Regulators of Immune Responses in Cancer. Cancers 2019, 11, 1037.
- Shaulian, E.; Karin, M. AP-1 as a regulator of cell life and death. Nat. Cell Biol. 2002, 4, E131–E136.
- Jochum, W.; Passegué, E.; Wagner, E.F. AP-1 in mouse development and tumorigenesis. Oncogene 2001, 20, 2401–2412.
- Rajkumar, S.V. Multiple myeloma: 2020 update on diagnosis, risk-stratification and management. Am. J. Hematol. 2020, 95, 548–567.
- Bianchi, G.; Munshi, N.C. Pathogenesis beyond the cancer clone(s) in multiple myeloma. Blood 2015, 125, 3049–3058.
- Manier, S.; Salem, K.Z.; Park, J.; Landau, D.A.; Getz, G.; Ghobrial, I.M. Genomic complexity of multiple myeloma and its clinical implications. Nat. Rev. Clin. Oncol. 2017, 14, 100–113.
- Morgan, G.J.; Walker, B.A.; Davies, F.E. The genetic architecture of multiple myeloma. Nat. Rev. Cancer 2012, 12, 335–348.
- Gasparetto, C.; Lentzsch, S.; Schiller, G.; Callander, N.; Tuchman, S.; Chen, C.; White, D.; Kotb, R.; Sutherland, H.; Sebag, M.; et al. Selinexor, daratumumab, and dexamethasone in patients with relapsed or refractory multiple myeloma. eJHaem 2021, 2, 56–65.
- Markham, A. Belantamab Mafodotin: First Approval. Drugs 2020, 80, 1607–1613.
- Kuehl, W.M.; Bergsagel, P.L. Multiple myeloma: Evolving genetic events and host interactions. Nat. Rev. Cancer 2002, 2, 175–187.
- Shapiro-Shelef, M.; Calame, K. Regulation of plasma-cell development. Nat. Rev. Immunol. 2005, 5, 230–242.
- Li, S.; Vallet, S.; Sacco, A.; Roccaro, A.; Lentzsch, S.; Podar, K. Targeting transcription factors in multiple myeloma: Evolving therapeutic strategies. Expert Opin. Investig. Drugs 2019, 28, 445–462.
- Pawlyn, C.; Morgan, G.J. Evolutionary biology of high-risk multiple myeloma. Nat. Rev. Cancer 2017, 17, 543–556.
- Bradner, J.E.; Hnisz, D.; Young, R.A. Transcriptional Addiction in Cancer. Cell 2017, 168, 629–643.
- Shaffer, A.L.; Emre, N.C.T.; Lamy, L.; Ngo, V.N.; Wright, G.; Xiao, W.; Powell, J.; Dave, S.; Yu, X.; Zhao, H.; et al. IRF4 addiction in multiple myeloma. Nature 2008, 454, 226–231.
- Holien, T.; Våtsveen, T.K.; Hella, H.; Waage, A.; Sundan, A. Addiction to c-MYC in multiple myeloma. Blood 2012, 120, 2450–2453.
- Jovanović, K.K.; Roche-Lestienne, C.; Ghobrial, I.M.; Facon, T.; Quesnel, B.; Manier, S. Targeting MYC in multiple myeloma. Leukemia 2018, 32, 1295–1306.
- Fan, F.; Podar, K. The Role of AP-1 Transcription Factors in Plasma Cell Biology and Multiple Myeloma Pathophysiology. Cancers 2021, 13, 2326. https://doi.org/10.3390/cancers13102326
- Ohkubo, Y.; Arima, M.; Arguni, E.; Okada, S.; Yamashita, K.; Asari, S.; Obata, S.; Sakamoto, A.; Hatano, M.; Wang, J.O.; et al. A Role for c- fos/Activator Protein 1 in B Lymphocyte Terminal Differentiation. J. Immunol. 2005, 174, 7703–7710.
- Grötsch, B.; Brachs, S.; Lang, C.; Luther, J.; Derer, A.; Schlötzer-Schrehardt, U.; Bozec, A.; Fillatreau, S.; Berberich, I.; Hobeika, E.; et al. The AP-1 transcription factor Fra1 inhibits follicular B cell differentiation into plasma cells. J. Exp. Med. 2014, 211, 2199–2212.
- Wagner, E.F. Bone development and inflammatory disease is regulated by AP-1 (Fos/Jun). Ann. Rheum. Dis. 2010, 69, i86–i88.
- Ubieta, K.; Garcia, M.; Grötsch, B.; Uebe, S.; Weber, G.F.; Stein, M.; Ekici, A.; Schett, G.; Mielenz, D.; Bozec, A. Fra-2 regulates B cell development by enhancing IRF4 and Foxo1 transcription. J. Exp. Med. 2017, 214, 2059–2071.
- Ise, W.; Kohyama, M.; Schraml, B.U.; Zhang, T.; Schwer, B.; Basu, U.; Alt, F.W.; Tang, J.; Oltz, E.M.; Murphy, T.L.; et al. The transcription factor BATF controls the global regulators of class-switch recombination in both B cells and T cells. Nat. Immunol. 2011, 12, 536–543.
- Dominguez-Sola, D.; Kung, J.; Holmes, A.B.; Wells, V.A.; Mo, T.; Basso, K.; Dalla-Favera, R. The FOXO1 Transcription Factor Instructs the Germinal Center Dark Zone Program. Immunity 2015, 43, 1064–1074.
- Ochiai, K.; Maienschein-Cline, M.; Simonetti, G.; Chen, J.; Rosenthal, R.; Brink, R.; Chong, A.S.; Klein, U.; Dinner, A.R.; Singh, H.; et al. Transcriptional Regulation of Germinal Center B and Plasma Cell Fates by Dynamical Control of IRF4. Immunity 2013, 38, 918–929.
- Agnarelli, A.; Chevassut, T.; Mancini, E.J. IRF4 in multiple myeloma—Biology, disease and therapeutic target. Leuk. Res. 2018, 72, 52–58.
- Annunziata, C.M.; Hernandez, L.; Davis, R.E.; Zingone, A.; Lamy, L.; Lam, L.T.; Hurt, E.M.; Shaffer, A.L.; Kuehl, W.M.; Staudt, L.M. A mechanistic rationale for MEK inhibitor therapy in myeloma based on blockade of MAF oncogene expression. Blood 2011, 117, 2396–2404.
- Hurt, E.M.; Wiestner, A.; Rosenwald, A.; Shaffer, A.; Campo, E.; Grogan, T.; Bergsagel, P.L.; Kuehl, W.M.; Staudt, L.M. Overexpression of c-maf is a frequent oncogenic event in multiple myeloma that promotes proliferation and pathological interactions with bone marrow stroma. Cancer Cell 2004, 5, 191–199.
- Suzuki, A.; Iida, S.; Kato-Uranishi, M.; Tajima, E.; Zhan, F.; Hanamura, I.; Huang, Y.; Ogura, T.; Takahashi, S.; Ueda, R.; et al. ARK5 is transcriptionally regulated by the Large-MAF family and mediates IGF-1-induced cell invasion in multiple myeloma: ARK5 as a new molecular determinant of malignant multiple myeloma. Oncogene 2005, 24, 6936–6944.
- Peterson, T.R.; Laplante, M.; Thoreen, C.C.; Sancak, Y.; Kang, S.A.; Kuehl, W.M.; Gray, N.S.; Sabatini, D.M. DEPTOR Is an mTOR Inhibitor Frequently Overexpressed in Multiple Myeloma Cells and Required for Their Survival. Cell 2009, 137, 873–886.
- Qiang, Y.; Ye, S.; Chen, Y.; Buros, A.F.; Edmonson, R.; van Rhee, F.; Barlogie, B.; Epstein, J.; Morgan, G.J.; Davies, F.E. MAF protein mediates innate resistance to proteasome inhibition therapy in multiple myeloma. Blood 2016, 128, 2919–2930.
- Qiang, Y.-W.; Ye, S.; Huang, Y.; Chen, Y.; Van Rhee, F.; Epstein, J.; Walker, B.A.; Morgan, G.J.; Davies, F.E. MAFb protein confers intrinsic resistance to proteasome inhibitors in multiple myeloma. BMC Cancer 2018, 18, 724.
- Miannay, B.; Minvielle, S.; Roux, O.; Drouin, P.; Avet-Loiseau, H.; Guérin-Charbonnel, C.; Gouraud, W.; Attal, M.; Facon, T.; Munshi, N.C.; et al. Logic programming reveals alteration of key transcription factors in multiple myeloma. Sci. Rep. 2017, 7, 9257.
- Podar, K.; Raab, M.S.; Tonon, G.; Sattler, M.; Barilà, D.; Zhang, J.; Tai, Y.-T.; Yasui, H.; Raje, N.; DePinho, R.A.; et al. Up-Regulation of c-Jun Inhibits Proliferation and Induces Apoptosis via Caspase-Triggered c-Abl Cleavage in Human Multiple Myeloma. Cancer Res. 2007, 67, 1680–1688.
- Fan, F.; Tonon, G.; Bashari, M.H.; Vallet, S.; Antonini, E.; Goldschmidt, H.; Schulze-Bergkamen, H.; Opferman, J.T.; Sattler, M.; Anderson, K.C.; et al. Targeting Mcl-1 for multiple myeloma (MM) therapy: Drug-induced generation of Mcl-1 fragment Mcl-1128-350 triggers MM cell death via c-Jun upregulation. Cancer Lett. 2014, 343, 286–294.
- Chen, L.; Wang, S.; Zhou, Y.; Wu, X.; Entin, I.; Epstein, J.; Yaccoby, S.; Xiong, W.; Barlogie, B.; Shaughnessy, J.D.; et al. Identification of early growth response protein 1 (EGR-1) as a novel target for JUN-induced apoptosis in multiple myeloma. Blood 2010, 115, 61–70.
- Saha, M.N.; Jiang, H.; Yang, Y.; Zhu, X.; Wang, X.; Schimmer, A.D.; Qiu, L.; Chang, H. Targeting p53 via JNK Pathway: A Novel Role of RITA for Apoptotic Signaling in Multiple Myeloma. PLoS ONE 2012, 7, e30215.
- Fan, F.; Bashari, M.H.; Morelli, E.; Tonon, G.; Malvestiti, S.; Vallet, S.; Jarahian, M.; Seckinger, A.; Hose, D.; Bakiri, L.; et al. The AP-1 transcription factor JunB is essential for multiple myeloma cell proliferation and drug resistance in the bone marrow microenvironment. Leukemia 2017, 31, 1570–1581.
- Fan, F.; Malvestiti, S.; Vallet, S.; Lind, J.; Garcia-Manteiga, J.M.; Morelli, E.; Jiang, Q.; Seckinger, A.; Hose, D.; Goldschmidt, H.; et al. JunB is a key regulator of multiple myeloma bone marrow angiogenesis. Leukemia 2021. https://doi.org/10.1038/s41375-021-01271-9; https://rdcu.be/ckOLI
- Grigoriadis, A.; Wang, Z.; Cecchini, M.; Hofstetter, W.; Felix, R.; Fleisch, H.; Wagner, E. c-Fos: A key regulator of osteoclast-macrophage lineage determination and bone remodeling. Science 1994, 266, 443–448.
- Matsuo, K.; Owens, J.M.; Tonko, M.; Elliott, C.; Chambers, T.J.; Wagner, E.F. Fosl1 is a transcriptional target of c-Fos during osteoclast differentiation. Nat. Genet. 2000, 24, 184–187.
- Takayanagi, H.; Kim, S.; Matsuo, K.; Suzuki, H.; Suzuki, T.; Sato, K.; Yokochi, T.; Oda, H.; Nakamura, K.; Ida, N.; et al. RANKL maintains bone homeostasis through c-Fos-dependent induction of interferon-β. Nature 2002, 416, 744–749.
- Takayanagi, H.; Kim, S.; Koga, T.; Nishina, H.; Isshiki, M.; Yoshida, H.; Saiura, A.; Isobe, M.; Yokochi, T.; Inoue, J.; et al. Induction and Activation of the Transcription Factor NFATc1 (NFAT2) Integrate RANKL Signaling in Terminal Differentiation of Osteoclasts. Dev. Cell 2002, 3, 889–901.
- Jochum, W.; David, J.-P.; Elliott, C.; Wutz, A.; Plenk, H.; Matsuo, K.; Wagner, E.F. Increased bone formation and osteosclerosis in mice overexpressing the transcription factor Fra-1. Nat. Med. 2000, 6, 980–984.
- Eferl, R.; Hoebertz, A.; Schilling, A.F.; Rath, M.; Karreth, F.; Kenner, L.; Amling, M.; Wagner, E.F. The Fos-related antigen Fra-1 is an activator of bone matrix formation. EMBO J. 2004, 23, 2789–2799.
- Bozec, A.; Bakiri, L.; Jimenez, M.; Schinke, T.; Amling, M.; Wagner, E.F. Fra-2/AP-1 controls bone formation by regulating osteoblast differentiation and collagen production. J. Cell Biol. 2010, 190, 1093–1106.
- Bozec, A.; Bakiri, L.; Hoebertz, A.; Eferl, R.; Schilling, A.F.; Komnenovic, V.; Scheuch, H.; Priemel, M.; Stewart, C.L.; Amling, M.; et al. Osteoclast size is controlled by Fra-2 through LIF/LIF-receptor signalling and hypoxia. Nature 2008, 454, 221–225.
- Kenner, L.; Hoebertz, A.; Beil, F.T.; Keon, N.; Karreth, F.; Eferl, R.; Scheuch, H.; Szremska, A.; Amling, M.; Schorpp-Kistner, M.; et al. Mice lacking JunB are osteopenic due to cell-autonomous osteoblast and osteoclast defects. J. Cell Biol. 2004, 164, 613–623.
- Ye, N.; Ding, Y.; Wild, C.; Shen, Q.; Zhou, J. Small Molecule Inhibitors Targeting Activator Protein 1 (AP-1). J. Med. Chem. 2014, 57, 6930–6948.
- Chen, A.; Koehler, A.N. Transcription Factor Inhibition: Lessons Learned and Emerging Targets. Trends Mol. Med. 2020, 26, 508–518.
- Dang, C.V.; Reddy, E.P.; Shokat, K.M.; Soucek, L. Drugging the “undruggable” cancer targets. Nat. Rev. Cancer 2017, 17, 502–508.
- Gonda, T.J.; Ramsay, R.G. Directly targeting transcriptional dysregulation in cancer. Nat. Rev. Cancer 2015, 15, 686–694.
- Bushweller, J.H. Targeting transcription factors in cancer—From undruggable to reality. Nat. Rev. Cancer 2019, 19, 611–624.
- Kim, E.; Ahuja, A.; Kim, M.-Y.; Cho, J.Y. DNA or Protein Methylation-Dependent Regulation of Activator Protein-1 Function. Cells 2021, 10, 461.
- Serafimova, I.M.; Pufall, M.A.; Krishnan, S.; Duda, K.; Cohen, M.S.; Maglathlin, R.L.; McFarland, J.M.; Miller, R.M.; Frödin, M.; Taunton, J. Reversible targeting of noncatalytic cysteines with chemically tuned electrophiles. Nat. Chem. Biol. 2012, 8, 471–476.
- Pellegrino, S.; Ronda, L.; Annoni, C.; Contini, A.; Erba, E.; Gelmi, M.L.; Piano, R.; Paredi, G.; Mozzarelli, A.; Bettati, S. Molecular insights into dimerization inhibition of c-Maf transcription factor. Biochim. Biophys. Acta Proteins Proteomics 2014, 1844, 2108–2115.
- Lathbridge, A.; Mason, J.M. Computational Competitive and Negative Design To Derive a Specific cJun Antagonist. Biochemistry 2018, 57, 6108–6118.
- Baxter, D.; Perry, S.R.; Hill, T.A.; Kok, W.M.; Zaccai, N.R.; Brady, R.L.; Fairlie, D.P.; Mason, J.M. Downsizing Proto-oncogene cFos to Short Helix-Constrained Peptides That Bind Jun. ACS Chem. Biol. 2017, 12, 2051–2061.
- Baxter, D.; Ullman, C.G.; Frigotto, L.; Mason, J.M. Exploiting Overlapping Advantages of In Vitro and In Cellulo Selection Systems to Isolate a Novel High-Affinity cJun Antagonist. ACS Chem. Biol. 2017, 12, 2579–2588.
- Pernelle, C.; Clerc, F.F.; Dureuil, C.; Bracco, L.; Tocque, B. An efficient screening assay for the rapid and precise determination of affinities between leucine zipper domains. Biochemistry 1993, 32, 11682–1168
- Worrall, J.A.R.; Mason, J.M. Thermodynamic analysis of Jun-Fos coiled coil peptide antagonists. FEBS J. 2011, 278, 663–672.
- Mason, J.M.; Schmitz, M.A.; Muller, K.M.; Arndt, K.M. Semirational design of Jun-Fos coiled coils with increased affinity: Universal implications for leucine zipper prediction and design. Proc. Natl. Acad. Sci. USA 2006, 103, 8989–8994.
- Bains, N.P.S.; Wilce, J.A.; Heuer, K.H.; Tunstall, M.; Mackey, J.P.; Bennett, M.R.; Weiss, A.S.; King, G.F. Zipping up transcription factors: Rational design of anti-Jun and anti-Fos peptides. Lett. Pept. Sci. 1997, 4, 67–77.
- Uchihashi, S.; Fukumoto, H.; Onoda, M.; Hayakawa, H.; Ikushiro, S.; Sakaki, T. Metabolism of the c-Fos/Activator Protein-1 Inhibitor T-5224 by Multiple Human UDP-Glucuronosyltransferase Isoforms. Drug Metab. Dispos. 2011, 39, 803–813.
- Izuta, S.; Ueki, M.; Ueno, M.; Nishina, K.; Shiozawa, S.; Maekawa, N. T-5224, a selective inhibitor of c-Fos/activator protein-1, attenuates lipopolysaccharide-induced liver injury in mice. Biotechnol. Lett. 2012, 34, 2175–2182.
- Dai, J.; Punchihewa, C.; Mistry, P.; Ooi, A.T.; Yang, D. Novel DNA bis-intercalation by MLN944, a potent clinical bisphenazine anticancer drug. J. Biol. Chem. 2004, 279, 46096–46103.
- Fanjul, A.; Dawson, M.I.; Hobbs, P.D.; Jong, L.; Cameron, J.F.; Harlev, E.; Graupner, G.; Lu, X.-P.; Pfahl, M. A new class of retinoids with selective inhibition of AP-1 inhibits proliferation. Nature 1994, 372, 107–111.
- Huang, C.; Ma, W.-Y.; Dawson, M.I.; Rincon, M.; Flavell, R.A.; Dong, Z. Blocking activator protein-1 activity, but not activating retinoic acid response element, is required for the antitumor promotion effect of retinoic acid. Proc. Natl. Acad. Sci. USA 1997, 94, 5826–5830.
- Olive, M.; Krylov, D.; Vinson, C.; Echlin, D.R.; Gardner, K.; Taparowsky, E. A Dominant Negative to Activation Protein-1 (AP1) That Abolishes DNA Binding and Inhibits Oncogenesis. J. Biol. Chem. 1997, 272, 18586–18594.
- He, W.; Wu, Y.; Tang, X.; Xia, Y.; He, G.; Min, Z.; Li, C.; Xiong, S.; Shi, Z.; Lu, Y.; et al. HDAC inhibitors suppress c-Jun/Fra-1-mediated proliferation through transcriptionally downregulating MKK7 and Raf1 in neuroblastoma cells. Oncotarget 2016, 7, 6727–6747.
- Kim, E.; Jang, J.; Park, J.G.; Kim, K.-H.; Yoon, K.; Yoo, B.C.; Cho, J.Y. Protein Arginine Methyltransferase 1 (PRMT1) Selective Inhibitor, TC-E 5003, Has Anti-Inflammatory Properties in TLR4 Signaling. Int. J. Mol. Sci. 2020, 21, 3058.
- Han, S.-S.; Keum, Y.-S.; Seo, H.-J.; Surh, Y.-J. Curcumin Suppresses Activation of NF-κB and AP-1 Induced by Phorbol Ester in Cultured Human Promyelocytic Leukemia Cells. J. Biochem. Mol. Biol. 2002, 35, 337–342.
- Boissy, P.; Andersen, T.L.; Abdallah, B.M.; Kassem, M.; Plesner, T.; Delaissé, J.-M. Resveratrol Inhibits Myeloma Cell Growth, Prevents Osteoclast Formation, and Promotes Osteoblast Differentiation. Cancer Res. 2005, 65, 9943–9952.
- Bai, F.; Liu, K.; Li, H.; Wang, J.; Zhu, J.; Hao, P.; Zhu, L.; Zhang, S.; Shan, L.; Ma, W.; et al. Veratramine modulates AP-1-dependent gene transcription by directly binding to programmable DNA. Nucleic Acids Res. 2018, 46, 546–557.