 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Eleni Gavriilaki | + 1629 word(s) | 1629 | 2020-06-03 07:55:35 | | | |
| 2 | Nicole Yin | -4 word(s) | 1625 | 2020-06-03 10:27:28 | | | | |
| 3 | Nicole Yin | -2 word(s) | 1623 | 2020-11-06 04:41:53 | | | | |
| 4 | Nicole Yin | -2 word(s) | 1623 | 2020-11-06 04:42:24 | | |
Video Upload Options
Immunotherapy with chimeric antigen receptor T (CAR-T cells) has been recently approved for patients with relapsed/refractory B-lymphoproliferative neoplasms. Along with great efficacy in patients with poor prognosis, CAR-T cells have been also linked with novel toxicities in a significant portion of patients. Cytokine release syndrome (CRS) and neurotoxicity present with unique clinical phenotypes that have not been previously observed. Nevertheless, they share similar characteristics with endothelial injury syndromes developing post hematopoietic cell transplantation (HCT). Evolution in complement therapeutics has attracted renewed interest in these life-threatening syndromes, primarily concerning transplant-associated thrombotic microangiopathy (TA-TMA). The immune system emerges as a key player not only mediating cytokine responses but potentially contributing to endothelial injury in CAR-T cell toxicity. The interplay between complement, endothelial dysfunction, hypercoagulability and inflammation seems to be a common denominator in these syndromes. As the indications for CAR-T cells and patient populations expand, there in an unmet clinical need of better understanding the pathophysiology of CAR-T cell toxicity. Therefore, this review aims to provide state-of-the art knowledge on: cellular therapies in clinical practice (indications and toxicities), endothelial injury syndromes and immunity and potential therapeutic targets.
1. Endothelial Injury Syndromes
Various endothelial injury syndromes result post allogeneic HCT, including transplant- associated thrombotic microangiopathy (TA-TMA), graft-versus-host disease (GVHD) and veno-occlusive disease/sinusoidal obstruction syndrome (SOS/VOD)[1].
TA-TMA is a life-threatening complication of HCT that manifests with microangiopathic hemolytic anemia, thrombocytopenia and often renal or neurologic dysfunction[2][3][4][5][6][7]. It is more common post allogeneic HCT, but has also been described post autologous HCT, especially in pediatric recipients[8]. Its diagnosis is largely hindered by the high incidence of cytopenias and organ dysfunction in HCT recipients. Indeed, renal and neurologic dysfunction are attributed to several causes post HCT, that are potentially life-threatening[9][10][11]. Endothelial injury has been long recognized as a contributor to the pathogenesis of TA-TMA. Various underlying processes (conditioning regimen toxicity, calcineurin inhibitors/CNIs, alloreactivity, bacterial products, and GVHD) contribute to a prothrombotic state, which may eventually lead to microvasculature thrombosis[12].
GVHD is the major cause of morbidity and mortality among allogeneic HCT survivors without relapse or secondary malignancy[13][14]. GVHD treatment consists mainly of immunosuppressive agents[15]. Prolonged immunosuppression is a risk factor of severe infections, leading to a vicious cycle of morbidity in GVHD patients[10][16][17]. Markers of endothelial dysfunction, such as endothelial microvesicles[18], are significantly increased 2–3 weeks post allogeneic HCT[19], as well as in patients with acute GVHD[20]. Endothelial activation has also been implicated in the pathophysiology of acute GVHD by a recent experimental study[21].
SOS/VOD disease of the liver has been traditionally considered a severe complication of allogeneic HCT, particularly in patients with known risk factors[22]. Although it manifests as a rare HCT complication thanks to advances in transplant modalities[23][24], calicheamicin-conjugated antibodies, gemtuzumab and inotuzumab ozogamicin, have led to renewed interest in this syndrome[25][26] [45,46]. Our group along with others has shown that changes in coagulation and fibrinolysis are predictive of SOS/VOD[27]. However, further studies have failed to identify useful biomarkers for routine clinical practice[22]. Its pathophysiology is strongly associated with damage observed in sinusoidal endothelial cells and in hepatocytes that continues with progressive venular occlusion[22].
Recent progress, mainly in the field of thrombotic microangiopathies (TMAs), has highlighted the role of complement as a common denominator in endothelial injury syndromes[28].
2. Immunity
The complement system is part of the immune system, comprising of more than 50 soluble and membrane-bound proteins[29]. It provides innate defense against microbes and mediating inflammatory responses. Except for inflammation, a link also exists between the complement system and platelet activation, leukocyte recruitment, endothelial cell activation and coagulation. Several reviews have tried to delineate the complex link between complement and thrombosis[30][31]. This link is basically established through interactions between C3, C5, and thrombin. Figure 1 summarizes complement activation and its interaction of complement with other pathways, that may implicate it in CAR-T cell toxicity.
The proximal complement cascade is activated by the classical, alternative, and lectin pathways. The classical pathway is mainly activated by antibody-antigen complexes recognized by complement component C1q[32]. This leads to the formation of classical pathway C3 convertase that cleaves C3, generating the anaphylatoxin C5a and C5 convertase. The latter cleaves C5 into C5a and C5b, initiating the terminal pathway of complement. In the terminal pathway, C5b binds to C6 and C7 generating C5b-7, that is able to insert into lipid layers of the membrane[33]. C5b-7 binds C8 and C9, forming a complex that unfolds in the membrane and binds several C9 molecules, thereby forming the membrane attack complex (MAC).
Interestingly, the alternative pathway of complement serves as an amplification loop for the lectin and classical pathways, accounting for roughly 80% of complement activation products[34]. The alternative pathway is continuously activated through slow spontaneous hydrolysis of C3, which forms C3(H2O)[35]. The activated C3(H2O) binds factor B, generating C3(H2O)B. Factor B is subsequently cleaved by factor D, generating the fluid phase APC C3 convertase, or C3(H2O)Bb. C3 convertase then catalyzes the cleavage of additional C3 molecules to generate C3a and C3b, which attach to cell surfaces[36]. This initiates the amplification loop, where C3b pairs with factor B on cell surfaces, bound factor B is cleaved by factor D to generate a second APC C3 convertase (C3bBb). Membrane-bound C3 convertase then cleaves additional C3 to generate more C3b deposits, closing the amplification loop. The binding and cleavage of an additional C3 molecule to C3 convertase forms the C5 convertase, initiating terminal pathway activation. Both C3 and C5 APC convertases are stabilized by properdin[37], which also serves as a selective pattern recognition molecule for de novo C3 APC convertase assembly[35]. Properdin is the only known positive regulator of complement. It increases the activity of C3 and C5 convertases, which amplify C3b deposition on cell surfaces[38].
Lectin pathway activation is initiated by mannose-binding lectins (MBLs)[39][40] and other pattern recognition molecules including ficolins and collectin 11[41]. These molecules act through MBL-associated serine proteases (MASPs), which generate the C3 convertase in a process similar to that of the classical pathway.
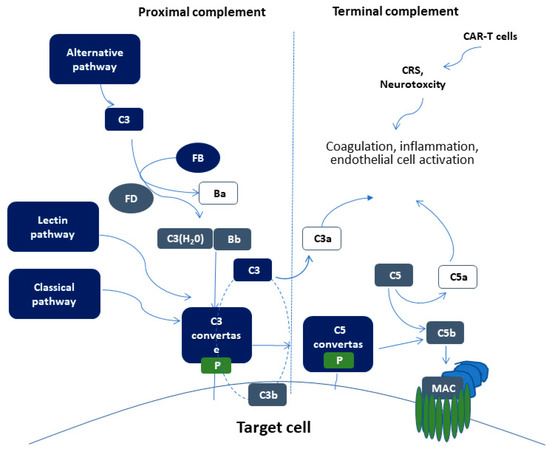
Figure 1. Schematic representation of complement activation. Proximal complement activation initiated by any of the three pathways (classical, alternative, or lectin pathway) leads to C3 activation and C3 convertase formation on C3-opsonized surfaces. C3 activation through the alternative pathway of complement amplifies this response (APC amplification loop, shown in dotted lines), culminating in pronounced C3 fragment deposition on target cells. In the presence of increased surface density of deposited C3b, the terminal (lytic) pathway is triggered, leading to membrane attack complex (MAC) formation on the surface of target cells. C3a and C5a mediate complement interactions with inflammation, coagulation, and endothelial cell activation. These alterations are also triggered by CAR (chimeric antigen receptor)-T cell toxicity syndromes, including CRS (cytokine release syndrome) and neurotoxicity.
3. Complement Activation in Endothelial Injury Syndromes
In TA-TMA, Jodele et al. first suggested that TA-TMA results from endothelial dysfunction after multiple triggers in genetically predisposed pediatric patients[7][42]. Initial data have shown excessive activation of terminal complement pathway through a rough marker of terminal complement activation, soluble C5b-9 levels[7]. Further studies have also confirmed complement activation on cell surface through functional assays[43]. Additionally, genomic data have suggested genetic susceptibility through rare mutations in complement-mediated genes[42]. Our group confirmed these data in adult patients[44], providing additional evidence of a vicious cycle of endothelial dysfunction, hypercoagulability, neutrophil and complement activation in TA-TMA[45]. A more recent study of transcriptome analysis in pediatric TA-TMA has shown activation of multiple complement pathways and an interplay between complement and interferon that perpetuates endothelial injury[46]. These data are in line with a previous clinical observation documenting complement-mediated TMA in patients with hemophagocytic lymphohistiocytosis (HLH), a rare clinical syndrome of excessive immune activation, characterized by signs and symptoms of extreme inflammation, largely driven by interferon γ and other pro-inflammatory cytokines[47].
Our understanding of the pathophysiology of TA-TMA has led to a revolution in therapeutics. Based on their success in patients with TMA and excessive complement activation[48][49], complement inhibitors have also shown success in TA-TMA. The first-in-class terminal complement inhibitor, eculizumab, has long been used in TA-TMA[50][51][52][53]. Real-world data suggest early initiation of treatment in patients with complement activation measured by soluble C5b-9 levels, as well as monitoring of treatment and dose adjustments yield better results[54]. Recently, narsoplimab (OMS721), a novel lectin pathway inhibitor targeting MASP-2 (mannan-binding lectin-associated serine protease-2), received breakthrough FDA designation, based on positive data in TA-TMA[55].
Clinical features of SOS/VOD share common characteristics with a syndrome observed during pregnancy, the HELLP (hemolysis, elevated liver enzymes, and low platelet number) syndrome. We and other groups have provided functional and genetic evidence pointing towards increased complement activation associated with complement-related germline mutations in patients with HELLP syndrome[56][57][58][59][60]. In this context, these syndromes resemble the disease model of complement-mediated hemolytic uremic syndrome (HUS)[28]. Different mutations in complement- related factors may lead to distinct phenotypes with similar characteristics as shown in other complement-related diseases, such as C3G-glomerupathy and age-related macular degeneration[61][62].
Earlier studies have suggested preliminary evidence of complement activation in patients with SOS/VOD. A subset of transplanted patients with SOS/VOD has shown increased complement activation markers at levels similar to those of patients with transplant-associated TMA. In addition, ADAMTS13 (A Disintegrin and Metalloproteinase with Thrombospondin motifs), a known regulator of TMAs, was reported lower in patients with SOS/VOD[63]. In line with these data, a previous case report documented increased complement activation in a SOS/VOD patient that was efficiently treated with the complement inhibitor C1 esterase inhibitor (C1-INH-C)[64]. Regarding genetic studies, Bucalossi et al. detected two complement factor H (CFH) variants in 3 SOS/VOD patients. Except for complement factor I (CFI), no other complement-related genes were studied[65].
References
- Enric Carreras; Maribel Diaz-Ricart; The role of the endothelium in the short-term complications of hematopoietic SCT. Bone Marrow Transplantation 2011, 46, 1495-1502, 10.1038/bmt.2011.65.
- Siribha Changsirikulchai; David Myerson; Katherine A. Guthrie; George B. McDonald; Charles E. Alpers; Sangeeta R. Hingorani; Renal Thrombotic Microangiopathy after Hematopoietic Cell Transplant: Role of GVHD in Pathogenesis. Clinical Journal of the American Society of Nephrology 2009, 4, 345-353, 10.2215/cjn.02070508.
- Hirohisa Nakamae; Takahisa Yamane; Taro Hasegawa; Mika Nakamae; Yoshiki Terada; Kiyoyuki Hagihara; Kensuke Ohta; Masayuki Hino; Risk factor analysis for thrombotic microangiopathy after reduced-intensity or myeloablative allogeneic hematopoietic stem cell transplantation. American Journal of Hematology 2006, 81, 525-531, 10.1002/ajh.20648.
- E Willems; Frédéric Baron; L Seidel; P Frère; G Fillet; Y Beguin; Comparison of thrombotic microangiopathy after allogeneic hematopoietic cell transplantation with high-dose or nonmyeloablative conditioning. Bone Marrow Transplantation 2009, 45, 689-693, 10.1038/bmt.2009.230.
- C Uderzo; Sonia Bonanomi; Alessandro Busca; Mila Renoldi; Pierantonio Ferrari; Massimo Iacobelli; Giuseppe Morreale; Edoardo Lanino; Claudio Annaloro; Aldo Della Volpe; Paolo Alessandrino; Daniela Longoni; Franco Locatelli; Haidi Sangalli; Attilio Rovelli; Risk Factors and Severe Outcome in Thrombotic Microangiopathy After Allogeneic Hematopoietic Stem Cell Transplantation. Transplantation 2006, 82, 638-644, 10.1097/01.tp.0000230373.82376.46.
- Vincent T. Ho; Corey Cutler; Shelly Carter; Paul Martin; Roberta Adams; Mary Horowitz; James Ferrara; Robert Soiffer; Sergio Giralt; Blood and Marrow Transplant Clinical Trials Network Toxicity Committee Consensus Summary: Thrombotic Microangiopathy after Hematopoietic Stem Cell Transplantation. Biology of Blood and Marrow Transplantation 2005, 11, 571-575, 10.1016/j.bbmt.2005.06.001.
- Sonata Jodele; Stella M. Davies; Adam Lane; Jane Khoury; Christopher E. Dandoy; Jens Goebel; Kasiani Myers; Michael Grimley; Jack Bleesing; Javier El-Bietar; Gregory Wallace; Ranjit S. Chima; Zachary Paff; Benjamin L. Laskin; Diagnostic and risk criteria for HSCT-associated thrombotic microangiopathy: a study in children and young adults. Blood 2014, 124, 645-653, 10.1182/blood-2014-03-564997.
- Michelle Schoettler; Leslie Lehmann; Anran Li; Clement Ma; Christine Duncan; Thrombotic Microangiopathy Following Pediatric Autologous Hematopoietic Cell Transplantation: A Report of Significant End-Organ Dysfunction in Eculizumab-Treated Survivors. Biology of Blood and Marrow Transplantation 2019, 25, e163-e168, 10.1016/j.bbmt.2018.12.840.
- Ioanna Sakellari; A Barbouti; G Bamichas; D Mallouri; P Kaloyannidis; S Fragidis; I Batsis; C Apostolou; A Karpouza; E Yannaki; C Smias; K Sombolos; Achilles Anagnostopoulos; GVHD-associated chronic kidney disease after allogeneic haematopoietic cell transplantation. Bone Marrow Transplantation 2013, 48, 1329-1334, 10.1038/bmt.2013.55.
- Maria Gavriilaki; Maria Mainou; Eleni Gavriilaki; Anna‐Bettina Haidich; Sotirios Papagiannopoulos; Ioanna Sakellari; Achilles Anagnostopoulos; Vasilis Kimiskidis; Neurologic complications after allogeneic transplantation: a meta-analysis.. Annals of Clinical and Translational Neurology 2019, 6, 2037-2047, 10.1002/acn3.50909.
- Ioanna Sakellari; Eleni Gavriilaki; Sotirios Papagiannopoulos; Maria Gavriilaki; Ioannis Batsis; Despina Mallouri; Anna Vardi; Varnavas Constantinou; Marianna Masmanidou; Evangelia Yannaki; Christos Smias; Triantafyllos Geroukis; Dimitrios Kazis; Vasileios Kimiskidis; Achilles Anagnostopoulos; Neurological adverse events post allogeneic hematopoietic cell transplantation: major determinants of morbidity and mortality. Journal of Neurology 2019, 266, 1960-1972, 10.1007/s00415-019-09372-3.
- Eleni Gavriilaki; Eugenia Gkaliagkousi; Savas Grigoriadis; Panagiota Anyfanti; Stella Douma; Achilles Anagnostopoulos; Hypertension in hematologic malignancies and hematopoietic cell transplantation: An emerging issue with the introduction of novel treatments.. Blood Reviews 2019, 35, 51-58, 10.1016/j.blre.2019.03.003.
- Eric J. Chow; Kenneth Wong; Stephanie J. Lee; Kara L. Cushing-Haugen; Mary E.D. Flowers; Debra L. Friedman; Wendy M. Leisenring; Paul J. Martin; Beth A. Mueller; K. Scott Baker; et al. Late Cardiovascular Complications after Hematopoietic Cell Transplantation. Biology of Blood and Marrow Transplantation 2014, 20, 794-800, 10.1016/j.bbmt.2014.02.012.
- Smita Bhatia; Liton Francisco; Andrea Carter; Can-Lan Sun; K. Scott Baker; James G. Gurney; Philip B. McGlave; Auayporn Nademanee; Margaret O'donnell; Norma K. C. Ramsay; et al.Leslie L. RobisonDavid SnyderAnthony SteinStephen J. FormanDaniel J. Weisdorf Late mortality after allogeneic hematopoietic cell transplantation and functional status of long-term survivors: report from the Bone Marrow Transplant Survivor Study. Blood 2007, 110, 3784-3792, 10.1182/blood-2007-03-082933.
- Ioanna Sakellari; Eleni Gavriilaki; Ioannis Batsis; Despina Mallouri; Alkistis-Kira Panteliadou; Andriana Lazaridou; Anna Vardi; Varnavas Constantinou; Evangelia Yannaki; Apostolia Papalexandri; et al.Panayotis KaloyannidisChristos SmiasAchilles Anagnostopoulos Favorable impact of extracorporeal photopheresis in acute and chronic graft versus host disease: Prospective single-center study. Journal of Clinical Apheresis 2018, 33, 654-660, 10.1002/jca.21660.
- Ioanna Sakellari; Eleni Gavriilaki; Maria Kaliou; Despina Mallouri; Ioannis Batsis; Evangelia Yannaki; Christos Smias; Damianos Sotiropoulos; Eleni Tsorlini; Achilles Anagnostopoulos; et al. Candida is an emerging pathogen beyond the neutropenic period of allogeneic hematopoietic cell transplantation. Clinical Transplantation 2017, 31, e12921, 10.1111/ctr.12921.
- D. Chatzidimitriou; Eleni Gavriilaki; Ioanna Sakellari; E. Diza; Hematopoietic cell transplantation and emerging viral infections. Journal of Medical Virology 2010, 82, 528-538, 10.1002/jmv.21696.
- Eugenia Gkaliagkousi; Eleni Gavriilaki; Ioannis Vasileiadis; Barbara Nikolaidou; Efthalia Yiannaki; Antonios Lazaridis; Areti Triantafyllou; Panagiota Anyfanti; Dimitra Markala; Ioannis Zarifis; et al.Stella Douma Endothelial Microvesicles Circulating in Peripheral and Coronary Circulation Are Associated With Central Blood Pressure in Coronary Artery Disease. American Journal of Hypertension 2019, 32, 1199-1205, 10.1093/ajh/hpz116.
- Shosaku Nomura; Kazuyoshi Ishii; Norihito Inami; Yutaka Kimura; Nobuhiko Uoshima; Hiroyuki Ishida; Takao Yoshihara; Fumiaki Urase; Yasuhiro Maeda; Kunio Hayashi; et al. Evaluation of Angiopoietins and Cell-Derived Microparticles after Stem Cell Transplantation. Biology of Blood and Marrow Transplantation 2008, 14, 766-774, 10.1016/j.bbmt.2008.04.005.
- Verena Pihusch; Andreas Rank; Ruth Steber; Markus Pihusch; Rudolf Pihusch; Bettina Toth; Erhard Hiller; Hans-Jochem Kolb; Endothelial Cell???Derived Microparticles in Allogeneic Hematopoietic Stem Cell Recipients. Transplantation 2006, 81, 1405-1409, 10.1097/01.tp.0000209218.24916.ba.
- Ran Zhang; Xiaoxiao Wang; Mei Hong; Ting Luo; Miaomiao Zhao; Haorui Shen; Jun Fang; Xiaojie Li; Sibin Zang; Ping Chen; et al.Dimin NiePeng ZhengQiuling WuLinghui Xia Endothelial microparticles delivering microRNA-155 into T lymphocytes are involved in the initiation of acute graft-versus-host disease following allogeneic hematopoietic stem cell transplantation. Oncotarget 2017, 8, 23360-23375, 10.18632/oncotarget.15579.
- Mohamad Mohty; F Malard; M Abecassis; E Aerts; A S Alaskar; M Aljurf; M Arat; P Bader; F Baron; Ali Bazarbachi; et al.D BlaiseF CiceriS CorbaciogluJ-H DalleR F DuarteT FukudaA HuynhTamas MassziM MichalletA NaglerM NichonghaileT PagluicaChristina PetersF B PetersenP G RichardsonT RuutuB N SavaniE WallhultIbrahim Yakoub-AghaE Carreras Sinusoidal obstruction syndrome/veno-occlusive disease: current situation and perspectives—a position statement from the European Society for Blood and Marrow Transplantation (EBMT). Bone Marrow Transplantation 2015, 50, 781-789, 10.1038/bmt.2015.52.
- Ioanna Sakellari; Ioannis Batsis; Zoi Bousiou; Despina Mallouri; Varnavas Constantinou; Eleni Gavriilaki; Christos Smias; Evangelia Yannaki; Panayotis Kaloyannidis; Giorgos Papaioannou; et al.Niki StavroyianniAntonia SyrigouDamianos SotiropoulosAsimina FylaktouAliki TsompanakouRiad SaloumAchilles Anagnostopoulos The Role of Low-dose Anti-thymocyte Globulin as Standard Prophylaxis in Mismatched and Matched Unrelated Hematopoietic Peripheral Stem Cell Transplantation for Hematologic Malignancies. Clinical Lymphoma Myeloma and Leukemia 2017, 17, 658-666, 10.1016/j.clml.2017.06.008.
- Ioanna Sakellari; Eleni Gavriilaki; Konstantinos Chatziioannou; Maria Papathanasiou; Despina Mallouri; Ioannis Batsis; Zoi Bousiou; Stella Bouziana; Varnavas Constantinou; Vassiliki Douka; et al.Chrysa ApostolouMichalis IskasChrysavgi LalayanniAnastasia AthanasiadouDamianos SotiropoulosEvangelia YannakiVasilis GianouzakosAchilles Anagnostopoulos Long-term outcomes of total body irradiation plus cyclophosphamide versus busulfan plus cyclophosphamide as conditioning regimen for acute lymphoblastic leukemia: a comparative study. Blut Zeitschrift für die gesamte Blutforschung 2018, 97, 1987-1994, 10.1007/s00277-018-3383-9.
- Juliette Lambert; Cécile Pautas; Christine Terré; Emmanuel Raffoux; Pascal Turlure; Denis Caillot; Ollivier Legrand; Xavier Thomas; Claude Gardin; Karïn Gogat-Marchant; et al.Stephen D. RubinRebecca J. BennerPierre BoussetClaude PreudhommeSylvie ChevretHerve DombretS Castaigne Gemtuzumab ozogamicin for de novo acute myeloid leukemia: final efficacy and safety updates from the open-label, phase III ALFA-0701 trial. Haematologica 2018, 104, 113-119, 10.3324/haematol.2018.188888.
- Elias J. Jabbour; Daniel J. DeAngelo; Matthias Stelljes; Wendy Stock; Michaela Liedtke; Nicola Gökbuget; Susan O'brien; Tao Wang; M. Luisa Paccagnella; Barbara Sleight; et al.Erik VandendriesAnjali S. AdvaniHagop M. Kantarjian Efficacy and safety analysis by age cohort of inotuzumab ozogamicin in patients with relapsed or refractory acute lymphoblastic leukemia enrolled in INO-VATE. Cancer 2018, 124, 1722-1732, 10.1002/cncr.31249.
- Ioannis Batsis; Evangelia Yannaki; Panayotis Kaloyannidis; Ioanna Sakellari; Christos Smias; Ioannis Georgoulis; Athanasios Fassas; Achilles Anagnostopoulos; Veno-occlusive disease prophylaxis with fresh frozen plasma and heparin in bone marrow transplantation. Thrombosis Research 2006, 118, 611-618, 10.1016/j.thromres.2005.10.010.
- Eleni Gavriilaki; Achilles Anagnostopoulos; Dimitrios C. Mastellos; Complement in Thrombotic Microangiopathies: Unraveling Ariadne's Thread Into the Labyrinth of Complement Therapeutics. Frontiers in Immunology 2019, 10, 337, 10.3389/fimmu.2019.00337.
- Eleni Gavriilaki; Robert A. Brodsky; Complementopathies and precision medicine. Journal of Clinical Investigation 2020, 130, 2152-2163, 10.1172/jci136094.
- Katerina Oikonomopoulou; Daniel Ricklin; Peter A. Ward; John D. Lambris; Interactions between coagulation and complement—their role in inflammation. Seminars in Immunopathology 2011, 34, 151-165, 10.1007/s00281-011-0280-x.
- Edward M. Conway; Complement-coagulation connections. Blood Coagulation & Fibrinolysis 2018, 29, 243-251, 10.1097/mbc.0000000000000720.
- Robert Brunhouse; John J. Cebra; Isotypes of IgG: Comparison of the primary structures of three pairs of isotypes which differ in their ability to activate complement. Molecular Immunology 1979, 16, 907-917, 10.1016/0161-5890(79)90089-0.
- K T Preissner; E R Podack; H J Müller-Eberhard; The membrane attack complex of complement: relation of C7 to the metastable membrane binding site of the intermediate complex C5b-7.. The Journal of Immunology 1985, 135, 445–451.
- Morten Harboe; Tom Eirik Mollnes; The alternative complement pathway revisited. Journal of Cellular and Molecular Medicine 2008, 12, 1074-1084, 10.1111/j.1582-4934.2008.00350.x.
- Claudio Cortes; Jennifer A. Ohtola; Gurpanna Saggu; Viviana Ferreira; Local release of properdin in the cellular microenvironment: role in pattern recognition and amplification of the alternative pathway of complement. Frontiers in Immunology 2013, 3, 412, 10.3389/fimmu.2012.00412.
- M K Pangburn; H J Müller-Eberhard; Initiation of the alternative complement pathway due to spontaneous hydrolysis of the thioester of C3.. Annals of the New York Academy of Sciences 1983, 421, 291–298.
- J. Chapitis; I H Lepow; Multiple sedimenting species of properdin in human serum and interaction of purified properdin with the third component of complement.. Journal of Experimental Medicine 1976, 143, 241-257, 10.1084/jem.143.2.241.
- Ferreira, V.P. Chapter 27—Properdin A2. In The Complement FactsBook, 2nd ed.; Barnum, S., Schein, T., Eds.; Academic Press: Cambridge, MA, USA, 2018; pp. 283–293.
- Olaf Neth; Dominic L. Jack; Alister W. Dodds; Helen Holzel; Nigel J. Klein; Malcolm W. Turner; Mannose-Binding Lectin Binds to a Range of Clinically Relevant Microorganisms and Promotes Complement Deposition. Infection and Immunity 2000, 68, 688-693.
- Mohammed Saifuddin; Gregory T. Spear; Yonghong Zhang; Henry Gewurz; Melanie L. Hart; Interaction of mannose-binding lectin with primary isolates of human immunodeficiency virus type 1. Journal of General Virology 2000, 81, 949-955, 10.1099/0022-1317-81-4-949.
- Dávid Héja; Andrea Kocsis; József Dobó; Katalin Szilágyi; Róbert Szász; Péter Závodszky; Gábor Pál; Péter Gál; Revised mechanism of complement lectin-pathway activation revealing the role of serine protease MASP-1 as the exclusive activator of MASP-2. Proceedings of the National Academy of Sciences 2012, 109, 10498-10503, 10.1073/pnas.1202588109.
- Sonata Jodele; Kejian Zhang; Fanggeng Zou; Benjamin Laskin; Christopher E. Dandoy; Kasiani C. Myers; Adam Lane; Jaroslav Meller; Mario Medvedovic; Jenny Chen; et al.Stella M. Davies The genetic fingerprint of susceptibility for transplant-associated thrombotic microangiopathy. Blood 2016, 127, 989-996, 10.1182/blood-2015-08-663435.
- Seth Rotz; Nathan Luebbering; Bradley Dixon; Eleni Gavriilaki; Robert A. Brodsky; Christopher E. Dandoy; Sonata Jodele; Stella M. Davies; In vitro evidence of complement activation in transplantation-associated thrombotic microangiopathy. Blood Advances 2017, 1, 1632-1634, 10.1182/bloodadvances.2017008250.
- Eleni Gavriilaki; Tasoula Touloumenidou; Ioanna Sakellari; Ioannis Batsis; Despina Mallouri; Fotis Psomopoulos; Maria Tsagiopoulou; Maria Koutra; Evangelia Yannaki; Apostolia Papalexandri; et al.Pat TaylorEmmanuel NikolousisMaria StamouliAndreas HolbroIoannis BaltadakisMaria LigaAlexandros SpyridonidisPanagiotis TsirigotisNikolaos CharchalakisDimitrios A. TsakirisRobert A. BrodskyJacob PasswegKostas StamatopoulosAchilles Anagnostopoulos Pretransplant Genetic Susceptibility: Clinical Relevance in Transplant-Associated Thrombotic Microangiopathy. Thrombosis and Haemostasis 2020, 120, 638-646, 10.1055/s-0040-1702225.
- Eleni Gavriilaki; Akrivi Chrysanthopoulou; Ioanna Sakellari; Ioannis Batsis; Despina Mallouri; Tasoula Touloumenidou; Apostolia Papalexandri; Alexandros Mitsios; Athanasios Arampatzioglou; Konstantinos Ritis; et al.Robert Alan BrodskyIoannis MitroulisAchilles Anagnostopoulos Linking Complement Activation, Coagulation, and Neutrophils in Transplant-Associated Thrombotic Microangiopathy.. Thrombosis and Haemostasis 2019, 119, 1433-1440, 10.1055/s-0039-1692721.
- Sonata Jodele; Mario Medvedovic; Nathan Luebbering; Jenny Chen; Christopher E. Dandoy; Benjamin L. Laskin; Stella M. Davies; Interferon-complement loop in transplant-associated thrombotic microangiopathy. Blood Advances 2020, 4, 1166-1177, 10.1182/bloodadvances.2020001515.
- Nicholas J. Gloude; Jack Bleesing; Stella M. Davies; Rebecca A Marsh; Michael B. Jordan; Sharat Chandra; Sonata Jodele; Thrombotic Microangiopathy Can Occur Before Transplant in Children with HLH. Biology of Blood and Marrow Transplantation 2017, 23, S233-S234, 10.1016/j.bbmt.2016.12.459.
- Christophe Legendre; C. Licht; P. Muus; L.A. Greenbaum; S. Babu; C. Bedrosian; C. Bingham; D.J. Cohen; Y. Delmas; K. Douglas; et al.F. EitnerT. FeldkampD. FouqueR.R. FurmanO. GaberM. HertheliusM. HourmantDiana KarpmanY. LebranchuC. MariatJ. MenneBruno Moulin’J. NürnbergerM. OgawaGiuseppe RemuzziT. RichardR. Sberro-SoussanB. SeverinoN.S. SheerinA. TrivelliL.B. ZimmerhacklT. GoodshipC. Loirat Terminal Complement Inhibitor Eculizumab in Atypical Hemolytic–Uremic Syndrome. New England Journal of Medicine 2013, 368, 2169-2181, 10.1056/nejmoa1208981.
- John Rathbone; Eva Kaltenthaler; Anna Richards; Paul Tappenden; Alice Bessey; Anna Cantrell; A systematic review of eculizumab for atypical haemolytic uraemic syndrome (aHUS). BMJ Open 2013, 3, e003573, 10.1136/bmjopen-2013-003573.
- Sonata Jodele; Christopher E. Dandoy; Lara Danziger-Isakov; Kasiani C. Myers; Javier El-Bietar; Adam Nelson; Gregory Wallace; Ashley Teusink-Cross; Stella M. Davies; Terminal Complement Blockade after Hematopoietic Stem Cell Transplantation Is Safe without Meningococcal Vaccination. Biology of Blood and Marrow Transplantation 2016, 22, 1337-1340, 10.1016/j.bbmt.2016.03.032.
- S Vasu; H Wu; A Satoskar; M Puto; J Roddy; W Blum; R Klisovic; L Andritsos; C Hofmeister; D M Benson; et al.Y EfeberaS JaglowskiS PenzaD CohenS DevineS Cataland Eculizumab therapy in adults with allogeneic hematopoietic cell transplant-associated thrombotic microangiopathy. Bone Marrow Transplantation 2016, 51, 1241-1244, 10.1038/bmt.2016.87.
- Flore Sicre De Fontbrune; Claire Galambrun; Anne Sirvent; Anne Huynh; Stanislas Faguer; Stephanie Nguyen; Jacques-Olivier Bay; Bénédicte Neven; Julie Moussi; Laurence Simon; et al.Aliénor XhaardMatthieu Resche-RigonAlix O’MearaVeronique Fremeaux-BacchiA. VeyradierGérard SociéP. CoppoRégis Peffaut De Latour Use of Eculizumab in Patients With Allogeneic Stem Cell Transplant-Associated Thrombotic Microangiopathy. Transplantation 2015, 99, 1953-1959, 10.1097/tp.0000000000000601.
- Stephan R. Bohl; Florian Kuchenbauer; Stefanie Von Harsdorf; Nadine Kloevekorn; Stefan S. Schönsteiner; Arefeh Rouhi; Phyllis Schwarzwälder; Hartmut Döhner; Donald Bunjes; Martin Bommer; et al. Thrombotic Microangiopathy after Allogeneic Stem Cell Transplantation: A Comparison of Eculizumab Therapy and Conventional Therapy. Biology of Blood and Marrow Transplantation 2017, 23, 2172-2177, 10.1016/j.bbmt.2017.08.019.
- Sonata Jodele; Christopher E. Dandoy; Adam Lane; Benjamin L Laskin; Ashley Teusink-Cross; Kasiani C Myers; Gregory H Wallace; Adam Nelson; Jack Bleesing; Ranjit S Chima; et al.Russel HirschThomas D. RyanStefanie Woolridge BenoitKana MizunoMikako WarrenStella M Davies Complement blockade for TA-TMA: lessons learned from large pediatric cohort treated with eculizumab. Blood 2020, null, null, 10.1182/blood.2019004218.
- Rambaldi, A.; Khaled, S.; Smith, M.; Zecca, M.; Kwong, Y.L.; Claes, K.; Leung, N.; Whitaker, S. Improved Survival Following OMS721 Treatment of Hematopoieic Stem Cell Transplant-associated Thrombotic Microangiopathy (HCT-TMA); EHA 2018: Stockholm, Sweden, 15 June 2018
- M Haeger; M Unander; A Bengtsson; Enhanced anaphylatoxin and terminal C5b-9 complement complex formation in patients with the syndrome of hemolysis, elevated liver enzymes, and low platelet count.. Obstetrics & Gynecology 1990, 76, , null.
- Richard M. Burwick; Raina N. Fichorova; Hassan Y. Dawood; Hidemi S. Yamamoto; Bruce B. Feinberg; Urinary Excretion of C5b-9 in Severe Preeclampsia. Hypertension 2013, 62, 1040-1045, 10.1161/hypertensionaha.113.01420.
- Arthur J. Vaught; Eleni Gavriilaki; Nancy Hueppchen; Karin Blakemore; Xuan Yuan; Sara M. Seifert; Sarah York; Robert A. Brodsky; Direct evidence of complement activation in HELLP syndrome: A link to atypical hemolytic uremic syndrome.. Experimental Hematology 2016, 44, 390-398, 10.1016/j.exphem.2016.01.005.
- Jane E. Salmon; Cara Heuser; Michael Triebwasser; M. Kathryn Liszewski; David Kavanagh; Lubka T. Roumenina; D. Ware Branch; Tim Goodship; Véronique Frémeaux-Bacchi; John P. Atkinson; Mutations in Complement Regulatory Proteins Predispose to Preeclampsia: A Genetic Analysis of the PROMISSE Cohort. PLoS Medicine 2011, 8, e1001013, 10.1371/journal.pmed.1001013.
- Arthur J. Vaught; Evan M. Braunstein; Jagar Jasem; Xuan Yuan; Igor Makhlin; Solange Eloundou; Andrea C. Baines; Samuel A. Merrill; Shruti Chaturvedi; Karin Blakemore; John Sperati; Robert A. Brodsky; Germline mutations in the alternative pathway of complement predispose to HELLP syndrome.. JCI Insight 2018, 3, , 10.1172/jci.insight.99128.
- Amy Osborne; Matteo Breno; Nicolo Ghiringhelli Borsa; Fengxiao Bu; Veronique Fremeaux-Bacchi; Daniel P. Gale; Lambertus P. Van Den Heuvel; David Kavanagh; Marina Noris; Sheila Pinto; et al.Pavithra M. RallapalliGiuseppe RemuzziSantiago Rodríguez De CordobaÁngela RuizRichard J.H. SmithPaula Vieira-MartinsElena VolokhinaValerie WilsonTimothy H. J. GoodshipStephen J. Perkins Statistical Validation of Rare Complement Variants Provides Insights into the Molecular Basis of Atypical Hemolytic Uremic Syndrome and C3 Glomerulopathy. The Journal of Immunology 2018, 200, 2464-2478, 10.4049/jimmunol.1701695.
- Maartje Geerlings; E.B. Volokhina; E.K. De Jong; N. Van De Kar; M. Pauper; C.B. Hoyng; L.P. Van Den Heuvel; Anneke I. Den Hollander; Genotype‐phenotype correlations of low‐frequency variants in the complement system in renal disease and age‐related macular degeneration. Clinical Genetics 2018, 94, 330-338, 10.1111/cge.13392.
- Jiaqian Qi; Jie Wang; Jia Chen; Jian Su; YaQiong Tang; Xiaojin Wu; Xiao Ma; Feng Chen; Changgeng Ruan; X. Long Zheng; et al.De-Pei WuYue Han Plasma levels of complement activation fragments C3b and sC5b-9 significantly increased in patients with thrombotic microangiopathy after allogeneic stem cell transplantation. Blut Zeitschrift für die gesamte Blutforschung 2017, 96, 1849-1855, 10.1007/s00277-017-3092-9.
- R Heying; W Nürnberger; U Spiekerkötter; U Göbel; Hepatic veno-occlusive disease with severe capillary leakage after peripheral stem cell transplantation: treatment with recombinant plasminogen activator and C1-esterase inhibitor concentrate. Bone Marrow Transplantation 1998, 21, 947-949, 10.1038/sj.bmt.1701211.
- Alessandro Bucalossi; Francesca Toraldo; Monica Tozzi; Mariapia Lenoci; Cinzia Castagnini; Rosangela Artuso; Alessandra Renieri; Giuseppe Marotta; Is complement alternative pathway disregulation involved in veno-occlusive disease of the liver?. Biology of Blood and Marrow Transplantation 2010, 16, 1749-1750, 10.1016/j.bbmt.2010.09.002.

