Immunotherapy with chimeric antigen receptor T (CAR-T cells) has been recently approved for patients with relapsed/refractory B-lymphoproliferative neoplasms. Along with great efficacy in patients with poor prognosis, CAR-T cells have been also linked with novel toxicities in a significant portion of patients. Cytokine release syndrome (CRS) and neurotoxicity present with unique clinical phenotypes that have not been previously observed. Nevertheless, they share similar characteristics with endothelial injury syndromes developing post hematopoietic cell transplantation (HCT). Evolution in complement therapeutics has attracted renewed interest in these life-threatening syndromes, primarily concerning transplant-associated thrombotic microangiopathy (TA-TMA). The immune system emerges as a key player not only mediating cytokine responses but potentially contributing to endothelial injury in CAR-T cell toxicity. The interplay between complement, endothelial dysfunction, hypercoagulability and inflammation seems to be a common denominator in these syndromes. As the indications for CAR-T cells and patient populations expand, there in an unmet clinical need of better understanding the pathophysiology of CAR-T cell toxicity. Therefore, this review aims to provide state-of-the art knowledge on: cellular therapies in clinical practice (indications and toxicities), endothelial injury syndromes and immunity and potential therapeutic targets.
- CAR-T
- endothelial injury syndrome
- toxicity
- complement
- immunity
1. Endothelial Injury Syndromes
Various endothelial injury syndromes result post allogeneic HCT, including transplant- associated thrombotic microangiopathy (TA-TMA), graft-versus-host disease (GVHD) and veno-occlusive disease/sinusoidal obstruction syndrome (SOS/VOD)[1] [21].
TA-TMA is a life-threatening complication of HCT that manifests with microangiopathic hemolytic anemia, thrombocytopenia and often renal or neurologic dysfunction[2][3][4][5][6][7] [22–27]. It is more common post allogeneic HCT, but has also been described post autologous HCT, especially in pediatric recipients[8] [28]. Its diagnosis is largely hindered by the high incidence of cytopenias and organ dysfunction in HCT recipients. Indeed, renal and neurologic dysfunction are attributed to several causes post HCT, that are potentially life-threatening[9][10][11] [29–31]. Endothelial injury has been long recognized as a contributor to the pathogenesis of TA-TMA. Various underlying processes (conditioning regimen toxicity, calcineurin inhibitors/CNIs, alloreactivity, bacterial products, and GVHD) contribute to a prothrombotic state, which may eventually lead to microvasculature thrombosis[12] [32].
GVHD is the major cause of morbidity and mortality among allogeneic HCT survivors without relapse or secondary malignancy[13][14] [33,34]. GVHD treatment consists mainly of immunosuppressive agents[15] [35]. Prolonged immunosuppression is a risk factor of severe infections, leading to a vicious cycle of morbidity in GVHD patients[10][16][17] [30,36,37]. Markers of endothelial dysfunction, such as endothelial microvesicles[18] [38], are significantly increased 2–3 weeks post allogeneic HCT[19] [39], as well as in patients with acute GVHD[20] [40]. Endothelial activation has also been implicated in the pathophysiology of acute GVHD by a recent experimental study[21] [41].
SOS/VOD disease of the liver has been traditionally considered a severe complication of allogeneic HCT, particularly in patients with known risk factors[22] [42]. Although it manifests as a rare HCT complication thanks to advances in transplant modalities[23][24] [43,44], calicheamicin-conjugated antibodies, gemtuzumab and inotuzumab ozogamicin, have led to renewed interest in this syndrome[25][26] [45,46]. Our group along with others has shown that changes in coagulation and fibrinolysis are predictive of SOS/VOD[27] [47]. However, further studies have failed to identify useful biomarkers for routine clinical practice[22] [42]. Its pathophysiology is strongly associated with damage observed in sinusoidal endothelial cells and in hepatocytes that continues with progressive venular occlusion[22] [42].
Recent progress, mainly in the field of thrombotic microangiopathies (TMAs), has highlighted the role of complement as a common denominator in endothelial injury syndromes[28] [48].
2. Immunity
The complement system is part of the immune system, comprising of more than 50 soluble and membrane-bound proteins[29] [49]. It provides innate defense against microbes and mediating inflammatory responses. Except for inflammation, a link also exists between the complement system and platelet activation, leukocyte recruitment, endothelial cell activation and coagulation. Several reviews have tried to delineate the complex link between complement and thrombosis[30][31] [50,51]. This link is basically established through interactions between C3, C5, and thrombin. Figure 1 summarizes complement activation and its interaction of complement with other pathways, that may implicate it in CAR-T cell toxicity.
The proximal complement cascade is activated by the classical, alternative, and lectin pathways. The classical pathway is mainly activated by antibody-antigen complexes recognized by complement component C1q[32] [52]. This leads to the formation of classical pathway C3 convertase that cleaves C3, generating the anaphylatoxin C5a and C5 convertase. The latter cleaves C5 into C5a and C5b, initiating the terminal pathway of complement. In the terminal pathway, C5b binds to C6 and C7 generating C5b-7, that is able to insert into lipid layers of the membrane[33] [53]. C5b-7 binds C8 and C9, forming a complex that unfolds in the membrane and binds several C9 molecules, thereby forming the membrane attack complex (MAC).
Interestingly, the alternative pathway of complement serves as an amplification loop for the lectin and classical pathways, accounting for roughly 80% of complement activation products[34] [54]. The alternative pathway is continuously activated through slow spontaneous hydrolysis of C3, which forms C3(H2O)[35] [55]. The activated C3(H2O) binds factor B, generating C3(H2O)B. Factor B is subsequently cleaved by factor D, generating the fluid phase APC C3 convertase, or C3(H2O)Bb. C3 convertase then catalyzes the cleavage of additional C3 molecules to generate C3a and C3b, which attach to cell surfaces[36] [56]. This initiates the amplification loop, where C3b pairs with factor B on cell surfaces, bound factor B is cleaved by factor D to generate a second APC C3 convertase (C3bBb). Membrane-bound C3 convertase then cleaves additional C3 to generate more C3b deposits, closing the amplification loop. The binding and cleavage of an additional C3 molecule to C3 convertase forms the C5 convertase, initiating terminal pathway activation. Both C3 and C5 APC convertases are stabilized by properdin[37] [57], which also serves as a selective pattern recognition molecule for de novo C3 APC convertase assembly[35] [55]. Properdin is the only known positive regulator of complement. It increases the activity of C3 and C5 convertases, which amplify C3b deposition on cell surfaces[38] [58].
Lectin pathway activation is initiated by mannose-binding lectins (MBLs)[39][40] [59,60] and other pattern recognition molecules including ficolins and collectin 11[41] [61]. These molecules act through MBL-associated serine proteases (MASPs), which generate the C3 convertase in a process similar to that of the classical pathway.
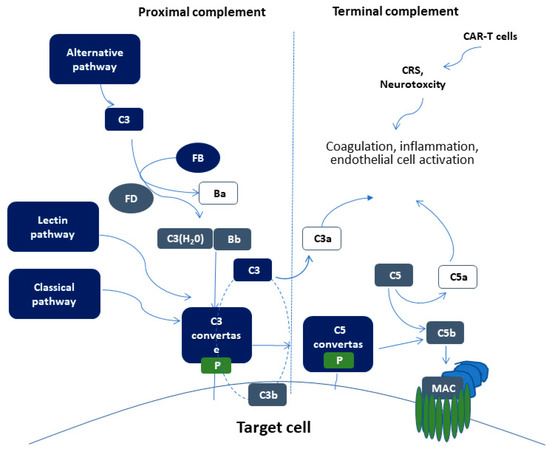
Figure 1. Schematic representation of complement activation. Proximal complement activation initiated by any of the three pathways (classical, alternative, or lectin pathway) leads to C3 activation and C3 convertase formation on C3-opsonized surfaces. C3 activation through the alternative pathway of complement amplifies this response (APC amplification loop, shown in dotted lines), culminating in pronounced C3 fragment deposition on target cells. In the presence of increased surface density of deposited C3b, the terminal (lytic) pathway is triggered, leading to membrane attack complex (MAC) formation on the surface of target cells. C3a and C5a mediate complement interactions with inflammation, coagulation, and endothelial cell activation. These alterations are also triggered by CAR (chimeric antigen receptor)-T cell toxicity syndromes, including CRS (cytokine release syndrome) and neurotoxicity.
3. Complement Activation in Endothelial Injury Syndromes
In TA-TMA, Jodele et al. first suggested that TA-TMA results from endothelial dysfunction after multiple triggers in genetically predisposed pediatric patients[7][42] [27,62]. Initial data have shown excessive activation of terminal complement pathway through a rough marker of terminal complement activation, soluble C5b-9 levels[7] [27]. Further studies have also confirmed complement activation on cell surface through functional assays[43] [63]. Additionally, genomic data have suggested genetic susceptibility through rare mutations in complement-mediated genes[42] [62]. Our group confirmed these data in adult patients[44] [64], providing additional evidence of a vicious cycle of endothelial dysfunction, hypercoagulability, neutrophil and complement activation in TA-TMA[45] [7]. A more recent study of transcriptome analysis in pediatric TA-TMA has shown activation of multiple complement pathways and an interplay between complement and interferon that perpetuates endothelial injury[46] [65]. These data are in line with a previous clinical observation documenting complement-mediated TMA in patients with hemophagocytic lymphohistiocytosis (HLH), a rare clinical syndrome of excessive immune activation, characterized by signs and symptoms of extreme inflammation, largely driven by interferon γ and other pro-inflammatory cytokines[47] [66].
Our understanding of the pathophysiology of TA-TMA has led to a revolution in therapeutics. Based on their success in patients with TMA and excessive complement activation[48][49] [67,68], complement inhibitors have also shown success in TA-TMA. The first-in-class terminal complement inhibitor, eculizumab, has long been used in TA-TMA[50][51][52][53] [69–72]. Real-world data suggest early initiation of treatment in patients with complement activation measured by soluble C5b-9 levels, as well as monitoring of treatment and dose adjustments yield better results[54] [73]. Recently, narsoplimab (OMS721), a novel lectin pathway inhibitor targeting MASP-2 (mannan-binding lectin-associated serine protease-2), received breakthrough FDA designation, based on positive data in TA-TMA[55] [74].
Clinical features of SOS/VOD share common characteristics with a syndrome observed during pregnancy, the HELLP (hemolysis, elevated liver enzymes, and low platelet number) syndrome. We and other groups have provided functional and genetic evidence pointing towards increased complement activation associated with complement-related germline mutations in patients with HELLP syndrome[56][57][58][59][60] [75–79]. In this context, these syndromes resemble the disease model of complement-mediated hemolytic uremic syndrome (HUS)[28] [48]. Different mutations in complement- related factors may lead to distinct phenotypes with similar characteristics as shown in other complement-related diseases, such as C3G-glomerupathy and age-related macular degeneration[61][62] [80,81].
Earlier studies have suggested preliminary evidence of complement activation in patients with SOS/VOD. A subset of transplanted patients with SOS/VOD has shown increased complement activation markers at levels similar to those of patients with transplant-associated TMA. In addition, ADAMTS13 (A Disintegrin and Metalloproteinase with Thrombospondin motifs), a known regulator of TMAs, was reported lower in patients with SOS/VOD[63] [82]. In line with these data, a previous case report documented increased complement activation in a SOS/VOD patient that was efficiently treated with the complement inhibitor C1 esterase inhibitor (C1-INH-C)[64] [83]. Regarding genetic studies, Bucalossi et al. detected two complement factor H (CFH) variants in 3 SOS/VOD patients. Except for complement factor I (CFI), no other complement-related genes were studied[65] [84].