 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Jean-baptiste Sortais | + 3361 word(s) | 3361 | 2021-05-07 09:50:58 | | | |
| 2 | Vivi Li | Meta information modification | 3361 | 2021-05-20 03:02:11 | | |
Video Upload Options
Hydrosilylation is a very versatile transformation consisting of the addition of a hydrosilane (H-SiR3) to an unsaturated bond. Organosilicon compounds have found widespread applications in our daily lives in silicon-based materials such as silicon rubbers, adhesives, paper release coating, and so forth. In addition, hydrosilylation is an atom economic reaction to access valuable organosilane intermediates for fine chemical synthesis
1. Introduction
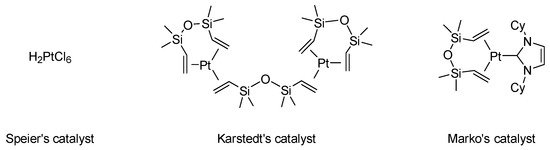
Hydrosilylation is a very versatile transformation consisting of the addition of a hydrosilane (H-SiR3) to an unsaturated bond. Organosilicon compounds have found widespread applications in our daily lives in silicon-based materials such as silicon rubbers, adhesives, paper release coating, and so forth. In addition, hydrosilylation is an atom economic reaction to access valuable organosilane intermediates for fine chemical synthesis [1][2]. For many years, platinum has been the metal of choice for designing hydrosilylation catalysts, Speier’s, Karstedt’s or Markó’s catalysts being the most representative examples (Figure 1). Other noble transition metals, such as rhodium, ruthenium, iridium or palladium, have also been used in this transformation [3]. However, the limited availability of these precious transition metals has prompted researchers to explore alternatives metals in the periodic table, in particular first row transition metals such as iron, cobalt and nickel [4][5][6].
In this regard, group 7 transition metals, manganese [7] and rhenium [8], were not an obvious choice at first glance. Indeed, rhenium complexes, in particular oxo-rhenium compounds, have been mainly recognized as efficient catalysts in oxidations, such as epoxidation or oxygen atom transfer reactions [9][10][11][12][13][14]. This is highlighted by organo-rhenium(VII) trioxide, particularly methyltrioxorhenium (MeReO3, abbreviated as MTO), arguably one of the most versatile transition metal catalysts known to date. Rhenium is also well-known to promote olefin metathesis [15] or aldehyde olefinations [16]. In complement, the coordination chemistry of rhenium was explored for the potential application of 186/188Re radioisotopes in nuclear medicine and bio-medical chemistry [17][18].
The application of rhenium derivatives in hydrosilylation [19][20] was not unveiled until 2003 [21][22]. Since the application of the lighter congener of rhenium, namely manganese, has been increasing lately in hydrosilylation [23][24], we report here the evolution of rhenium in this catalytic transformation.
2. Hydrosilylation of C=O, C=N, C≡N and NO2 Bonds
2.1. Hydrosilylation of Carbonyl Derivatives
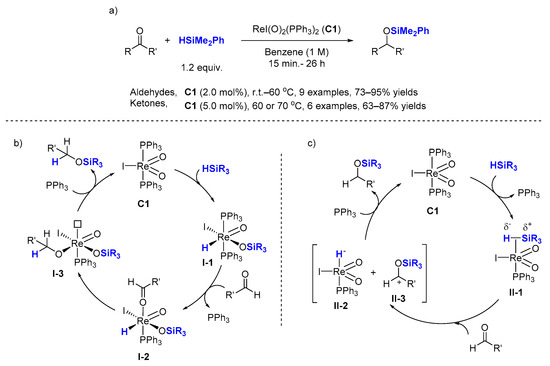
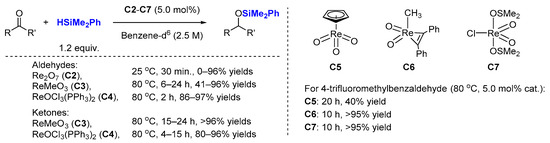
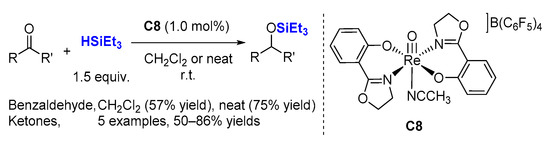
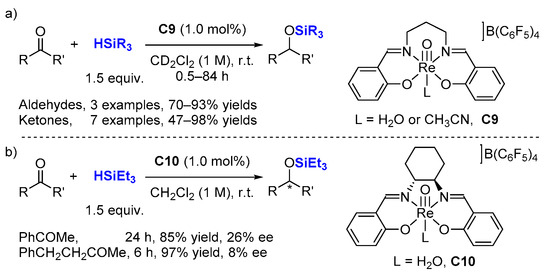
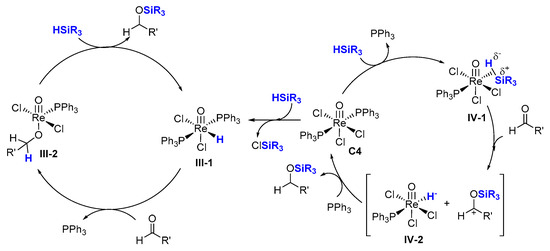
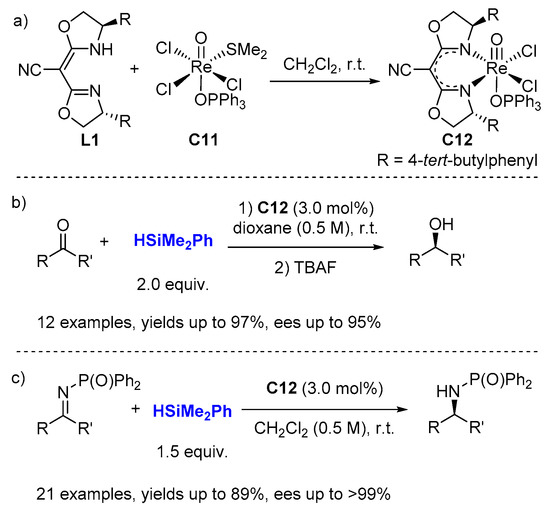
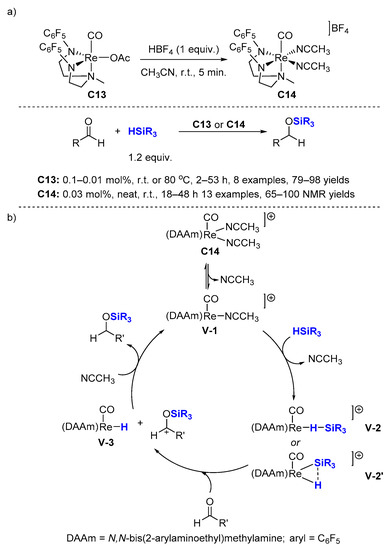
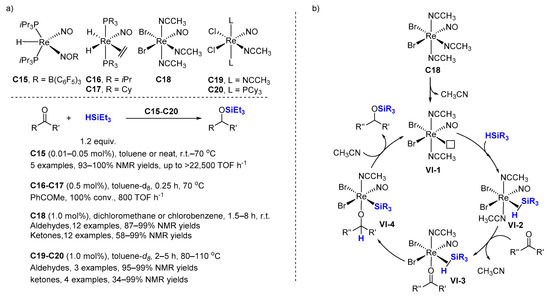
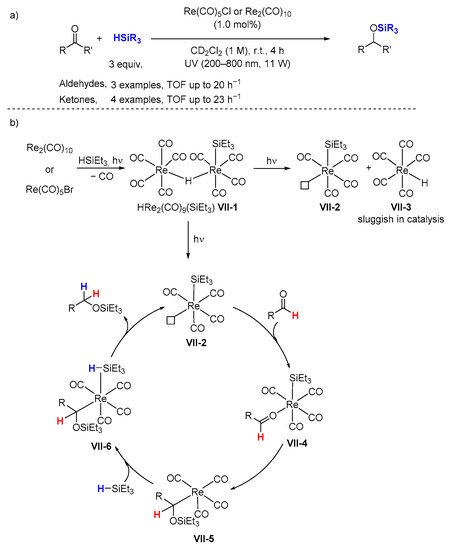
2.2. Hydrosilylation of Nitriles
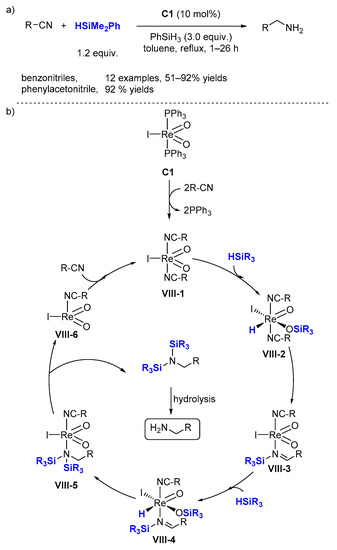
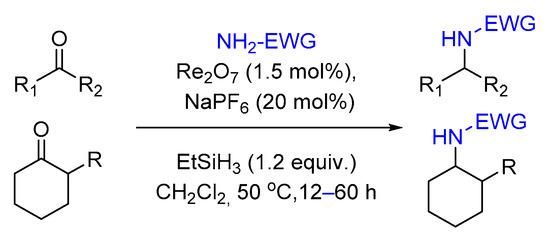
2.3. Reductive Amination
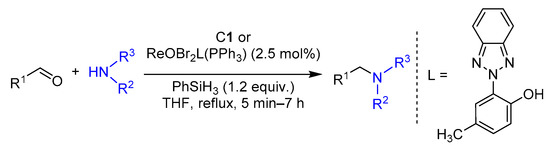
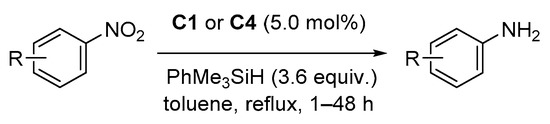
2.4. Hydrosilylation of Nitro Derivatives
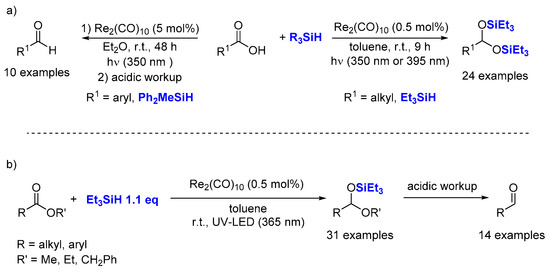
2.5. Hydrosilylation of Carboxyl Acids Derivatives
3. Hydrosilylation of C=C Bonds
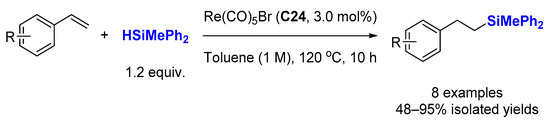
3.1. Hydrosilylation of Alkenes
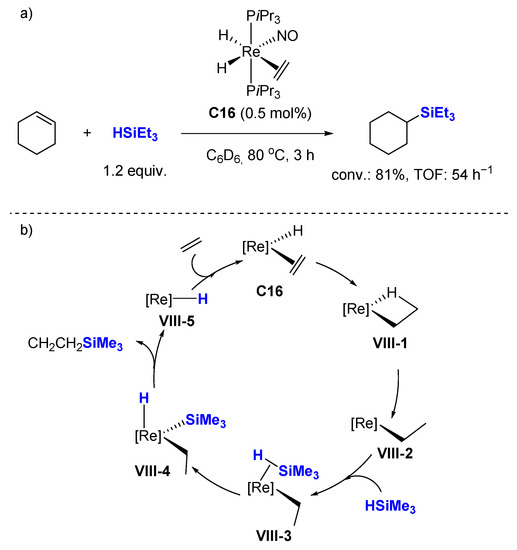
3.2. Dehydrogenative Silylation of Alkenes
Dehydrogenative silylation is often considered an undesired side reaction in the hydrosilylation of alkenes, but the selective production of vinylsilanes is also a valuable transformation. In this context; Berke et al. reported several rhenium mono-nitrosyl complexes (C18 [60], C19–C20 [47], C25–C26 [61], C27–C28 [62]) catalyzing the dehydrogenative silylation of alkenes with high chemoselectivity to produce silyl alkenes, along with the corresponding alkanes, with 1.0–4.0 mol% cat., at 70–110 °C in toluene-d8 (Scheme 18). Hydrosilylation products appear only rarely in very minor amounts. The complexes C19–C20 and C27–C28 gave better E/Z selectivity (96:4→99:1). Noticeably, ethylene can be transformed quantitatively into the corresponding dehydrogenative silylation product with C27–C28 as catalysts with a ratio dehydro/hydro silylation products of 79:21 and 68:32, respectively. C18 catalyzes the dehydrogenative coupling of styrene derivatives with HSiMe2Ph leading to dehydro/hydro silylation products in ratio of about 85/15.
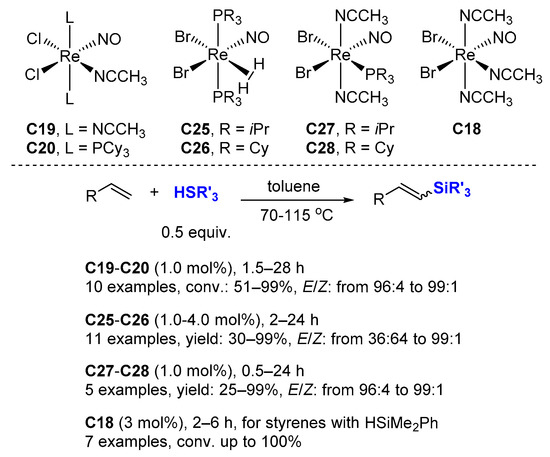
Scheme 18. Rhenium mono-nitrosyl complexes catalyzed dehydrogenative silylation of alkenes.
References
- Marciniec, B. Catalysis of hydrosilylation of carbon-carbon multiple bonds: Recent progress. Silicon Chem. 2002, 1, 155–174.
- Roy, A.K. A Review of Recent Progress in Catalyzed Homogeneous Hydrosilation (Hydrosilylation). Adv. Organomet. Chem. 2007, 55, 1–59.
- Nakajima, Y.; Shimada, S. Hydrosilylation reaction of olefins: Recent advances and perspectives. RSC Adv. 2015, 5, 20603–20616.
- Tamang, S.R.; Findlater, M. Emergence and Applications of Base Metals (Fe, Co, and Ni) in Hydroboration and Hydrosilylation. Molecules 2019, 24, 3194.
- Du, X.; Huang, Z. Advances in Base-Metal-Catalyzed Alkene Hydrosilylation. ACS Catal. 2017, 7, 1227–1243.
- Obligacion, J.V.; Chirik, P.J. Earth-abundant transition metal catalysts for alkene hydrosilylation and hydroboration. Nat. Rev. Chem. 2018, 2, 15–34.
- Valyaev, D.A.; Lavigne, G.; Lugan, N. Manganese organometallic compounds in homogeneous catalysis: Past, present, and prospects. Coord. Chem. Rev. 2016, 308, 191–235.
- Kuninobu, Y.; Takai, K. Organic Reactions Catalyzed by Rhenium Carbonyl Complexes. Chem. Rev. 2011, 111, 1938–1953.
- Harms, R.G.; Herrmann, W.A.; Kühn, F.E. Organorhenium dioxides as oxygen transfer systems: Synthesis, reactivity, and applications. Coord. Chem. Rev. 2015, 296, 1–23.
- Owens, G.S.; Arias, J.; Abu-Omar, M.M. ChemInform Abstract: Rhenium Oxo Complexes in Catalytic Oxidations. ChemInform 2010, 31, 317–363.
- Romão, C.C.; Kühn, F.E.; Herrmann, W.A. Rhenium(VII) Oxo and Imido Complexes: Synthesis, Structures, and Applications. Chem. Rev. 1997, 97, 3197–3246.
- Espenson, J.H. Atom-transfer reactions catalyzed by methyltrioxorhenium(VII)-mechanisms and applications. Chem. Commun. 1999, 6, 479–488.
- Fuchs, P.L. Catalytic Oxidation Reagents; John Wiley & Sons: Chichester, UK, 2013.
- Yudin, A.K.; Sharpless, K.B. Bis(trimethylsilyl) Peroxide Extends the Range of Oxorhenium Catalysts for Olefin Epoxidation. J. Am. Chem. Soc. 1997, 119, 11536–11537.
- Herrmann, W.A.; Kuchler, J.G.; Felixberger, J.K.; Herdtweck, E.; Wagner, W. Methylrhenium Oxides: Synthesis from R2O7 and Catalytic Activity in Olefin Metathesis. Angew. Chem. Int. Ed. 1988, 27, 394–396.
- Herrmann, W.A.; Roesky, P.W.; Wang, M.; Scherer, W. Multiple Bonds between Main-Group Elements and Transition Metals. 135. Oxorhenium(V) Catalysts for the Olefination of Aldehydes. Organometallics 1994, 13, 4531–4535.
- Volkert, W.A.; Hoffman, T.J. Therapeutic radiopharmaceuticals. Chem. Rev. 1999, 99, 2269–2292.
- Dilworth, J.R.; Parrott, S.J. The biomedical chemistry of technetium and rhenium. Chem. Soc. Rev. 1998, 27, 43–55.
- Du, G.; Abu-Omar, M.M. ChemInform Abstract: Oxo and Imido Complexes of Rhenium and Molybdenum in Catalytic Reductions. Chemistry 2009, 40, 1185–1198.
- Kobayashi, Y. Reduction with Hydrosilanes Catalyzed by Metal-oxo Complexes. J. Synth. Org. Chem. Jpn. 2010, 68, 866–867.
- Kennedy-Smith, J.J.; Nolin, K.A.; Gunterman, H.P.; Toste, F.D. Reversing the Role of the Metal-Oxygen π-Bond. Chemose-lective Catalytic Reductions with a Rhenium(V)-Dioxo Complex. J. Am. Chem. Soc. 2003, 125, 4056–4057.
- Thiel, W.R. On the Way to a New Class of Catalysts—High-Valent Transition-Metal Complexes That Catalyze Reductions. Angew. Chem. Int. Ed. 2003, 42, 5390–5392.
- Yang, X.; Wang, C. Manganese-Catalyzed Hydrosilylation Reactions. Chem. Asian J. 2018, 13, 2307–2315.
- Royo, B. Recent advances in catalytic hydrosilylation of carbonyl groups mediated by well-defined first-row late transition metals. Adv. Organomet. Chem. 2019, 72, 59–102.
- Nolin, K.A.; Krumper, J.R.; Pluth, M.D.; Bergman, A.R.G.; Toste, F.D. Analysis of an Unprecedented Mechanism for the Catalytic Hydrosilylation of Carbonyl Compounds. J. Am. Chem. Soc. 2007, 129, 14684–14696.
- Chung, L.W.; Lee, H.G.; Lin, Z.; Wu, Y.-D. Computational Study on the Reaction Mechanism of Hydrosilylation of Carbonyls Catalyzed by High-Valent Rhenium(V)−Di-oxo Complexes. J. Org. Chem. 2006, 71, 6000–6009.
- Huang, L.; Wang, W.; Wei, X.; Wei, H. New Insights into Hydrosilylation of Unsaturated Carbon-Heteroatom (C=O, C=N) Bonds by Rhenium(V)-Dioxo Complexes. J. Phys. Chem. A 2015, 119, 3789–3799.
- Royo, B.; Romão, C.C. Reduction of carbonyl groups by high-valent rhenium oxides. J. Mol. Catal. A Chem. 2005, 236, 107–112.
- Ison, E.A.; Trivedi, E.R.; Corbin, R.A.; Abu-Omar, M.M. Mechanism for Reduction Catalysis by Metal Oxo: Hydrosilation of Organic Carbonyl Groups Catalyzed by a Rhenium(V) Oxo Complex. J. Am. Chem. Soc. 2005, 127, 15374–15375.
- Ison, E.A.; Corbin, R.A.; Abu-Omar, M.M. Hydrogen Production from Hydrolytic Oxidation of Organosilanes Using a Cationic Oxorhenium Catalyst. J. Am. Chem. Soc. 2005, 127, 11938–11939.
- Du, G.; Abu-Omar, M.M. Catalytic Hydrosilylation of Carbonyl Compounds with Cationic Oxorhenium(V) Salen. Organometallics 2006, 25, 4920–4923.
- Ison, E.A.; Cessarich, J.E.; Du, G.; Fanwick, A.P.E.; Abu-Omar, M.M. Synthesis of Cationic Oxorhenium Salen Complexes via μ-Oxo Abstraction and Their Activity in Catalytic Reductions. Inorg. Chem. 2006, 45, 2385–2387.
- Du, G.; Fanwick, P.E.; Abu-Omar, M.M. Cationic oxorhenium chiral salen complexes for asymmetric hydrosilylation and kinetic resolution of alcohols. Inorg. Chim. Acta 2008, 361, 3184–3192.
- Du, G.; Fanwick, P.E.; Abu-Omar, M.M. Mechanistic Insight into Hydrosilylation Reactions Catalyzed by High Valent Re⋮X (X = O, NAr, or N) Complexes: The Silane (SiH) Does Not Add across the Metal−Ligand Multiple Bond. J. Am. Chem. Soc. 2007, 129, 5180–5187.
- Gu, P.; Wang, W.; Wang, Y.; Wei, H. Hydrosilylation of Carbonyls Catalyzed by the Rhenium(V) Oxo Complex [Re(O)(hoz)2]+—A Non-Hydride Pathway. Organometallics 2012, 32, 47–51.
- Huang, L.; Zhang, Y.; Wei, H. Role of the Isolable Hydride Intermediate in the Hydrosilylation of Carbonyl Compounds Catalyzed by the High-Valent Mono-Oxido-Rhenium(V) Complex. Eur. J. Inorg. Chem. 2014, 2014, 5714–5723.
- Huang, L.; Wang, W.; Wei, H. A computational study on high-valent mono-oxo-rhenium(V) complex-catalyzed hydrosilyla-tion of carbonyls: What a difference an oxo ligand makes. J. Mol. Catal. A Chem. 2015, 400, 31–41.
- Nolin, K.A.; Ahn, R.W.; Kobayashi, Y.; Kennedy-Smith, J.J.; Toste, F.D. Enantioselective Reduction of Ketones and Imines Catalyzed by (CN-Box)ReV-Oxo Complexes. Chem. Eur. J. 2010, 16, 9555–9562.
- Nolin, K.A.; Ahn, R.W.; Toste, F.D. Enantioselective reduction of imines catalyzed by a Rhenium (V)− Oxo complex. J. Am. Chem. Soc. 2005, 127, 12462–12463.
- Wang, J.; Wang, W.; Huang, L.; Yang, X.; Wei, H. The Unexpected Mechanism Underlying the High-Valent Mono-Oxo-Rhenium(V) Hydride Catalyzed Hydrosilylation of C=N Functionalities: Insights from a DFT Study. ChemPhysChem 2015, 16, 1052–1060.
- Smeltz, J.L.; Boyle, P.D.; Ison, E.A. Role of Low-Valent Rhenium Species in Catalytic Hydrosilylation Reactions with Ox-orhenium Catalysts. Organometallics 2012, 31, 5994–5997.
- Pérez, D.E.; Smeltz, J.L.; Sommer, R.D.; Boyle, P.D.; Ison, E.A. Cationic rhenium(iii) complexes: Synthesis, characterization, and reactivity for hydrosilylation of aldehydes. Dalton Trans. 2017, 46, 4609–4616.
- Brown, C.A.; Abrahamse, M.; Ison, E.A. Re–Silane complexes as frustrated lewis pairs for catalytic hydrosilylation. Dalton Trans. 2020, 49, 11403–11411.
- Huang, W.; Berke, H. Rhenium Complexes as Highly Active Catalysts for the Hydrosilylation of Carbonyl Compounds. Chim. Int. J. Chem. 2005, 59, 113–115.
- Choualeb, A.; Maccaroni, E.; Blacque, O.; Schmalle, H.W.; Berke, H. Rhenium Nitrosyl Complexes for Hydrogenations and Hydrosilylations. Organometallics 2008, 27, 3474–3481.
- Dong, H.; Berke, H. A Convenient and Efficient Rhenium-Catalyzed Hydrosilylation of Ketones and Aldehydes. Adv. Synth. Catal. 2009, 351, 1783–1788.
- Jiang, Y.; Blacque, O.; Berke, H. Probing the catalytic potential of chloro nitrosyl rhenium(i) complexes. Dalton Trans. 2011, 40, 2578–2587.
- Toh, C.K.; Sum, Y.N.; Fong, W.K.; Ang, S.G.; Fan, W.Y. Catalytic Hydrosilylation of Carbonyls via Re(CO)5Cl Photolysis. Organometallics 2012, 31, 3880–3887.
- Cabrita, I.; Fernandes, A.C. A novel efficient and chemoselective method for the reduction of nitriles using the system silane/oxo-rhenium complexes. Tetrahedron 2011, 67, 8183–8186.
- Li, B.; Sortais, J.-B.; Darcel, C. Amine synthesis via transition metal homogeneous catalysed hydrosilylation. RSC Adv. 2016, 6, 57603–57625.
- Sousa, S.C.; Fernandes, A.C. Efficient and highly chemoselective direct reductive amination of aldehydes using the system silane/oxorhenium complexes. Adv. Synth. Catal. 2010, 352, 2218–2226.
- Bernardo, J.R.; Sousa, S.C.A.; Florindo, P.R.; Wolff, M.; Machura, B.; Fernandes, A.C. Efficient and chemoselective direct reductive amination of aromatic aldehydes catalyzed by oxo–rhenium complexes containing heterocyclic ligands. Tetrahedron 2013, 69, 9145–9154.
- Das, B.G.; Ghorai, P. Stereoselective direct reductive amination of ketones with electron-deficient amines using Re2O7/NaPF6 catalyst. Org. Biomol. Chem. 2013, 11, 4379–4382.
- de Noronha, R.G.; Romão, C.C.; Fernandes, A.C. Highly Chemo- and Regioselective Reduction of Aromatic Nitro Compounds Using the System Silane/Oxo-Rhenium Complexes. J. Org. Chem. 2009, 74, 6960–6964.
- Zheng, J.; Chevance, S.; Darcel, C.; Sortais, J.-B. Selective reduction of carboxylic acids to aldehydes through manganese catalysed hydrosilylation. Chem. Commun. 2013, 49, 10010–10012.
- Wei, D.; Buhaibeh, R.; Canac, Y.; Sortais, J.-B. Rhenium-Catalyzed Reduction of Carboxylic Acids with Hydrosilanes. Org. Lett. 2019, 21, 7713–7716.
- Wei, D.; Buhaibeh, R.; Canac, Y.; Sortais, J.-B. Manganese and rhenium-catalyzed selective reduction of esters to aldehydes with hydrosilanes. Chem. Commun. 2020, 56, 11617–11620.
- Zhao, W.-G.; Hua, R. Highly regioselective rhenium-catalyzed hydrosilylation of styrenes. Eur. J. Org. Chem. 2006, 5495–5498.
- Liu, L.; Bi, S.; Sun, M.; Yuan, X.; Zheng, N.; Li, P. Mechanistic investigation on hydrogenation and hydrosilylation of ethylene catalyzed by rhenium nitrosyl complex. J. Organomet. Chem. 2009, 694, 3343–3348.
- Dong, H.; Jiang, Y.; Berke, H. Rhenium-mediated dehydrogenative silylation and highly regioselective hydrosilylation of nitrile substituted olefins. J. Organomet. Chem. 2014, 750, 17–22.
- Jiang, Y.; Blacque, O.; Fox, T.; Frech, C.M.; Berke, H. Highly selective dehydrogenative silylation of alkenes catalyzed by rhenium complexes. Chem. Eur. J. 2009, 15, 2121–2128.
- Jiang, Y.; Blacque, O.; Fox, T.; Frech, C.M.; Berke, H. Facile Synthetic Access to Rhenium(II) Complexes: Activation of Carbon-Bromine Bonds by Single-Electron Transfer. Chem. A Eur. J. 2010, 16, 2240–2249.



