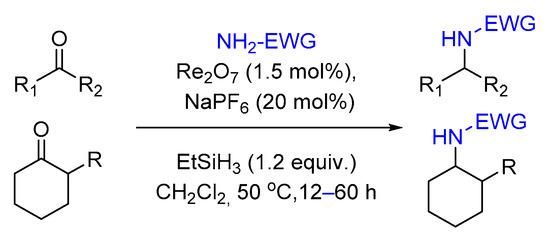
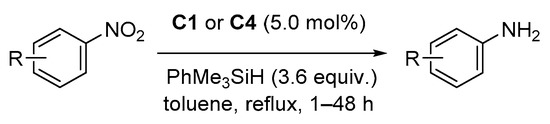
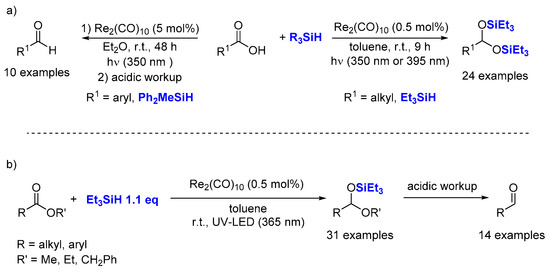
2.1. Hydrosilylation of Carbonyl Derivatives
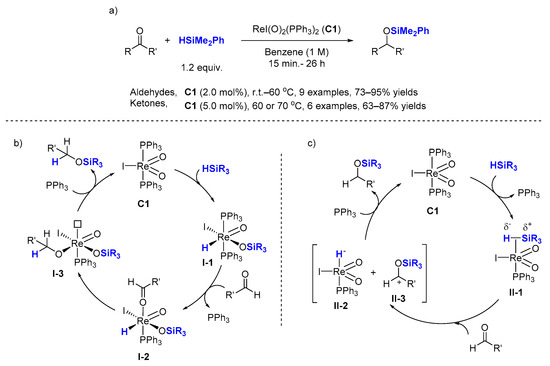
The first rhenium-catalyzed hydrosilylation was described in 2003 by Toste et al. (
Scheme 1a). This seminal contribution represents the first example of a hydrosilylation catalyst with a high-valent metal bearing two terminal oxo ligands, reversing the traditional use of metal-oxo complexes in catalysis. Indeed, the hydrosilylation of aldehydes and ketones was promoted by the readily available iododioxo(bistriphenylphosphine)rhenium(V) complex [(PPh
3)
2Re(O)
2I] (
C1) [
21,
22]. The scope of this reaction included aromatic or aliphatic ketones and aldehydes with a good tolerance of functional groups (amino, nitro, halo, ester, cyano, cyclopropyl and alkene groups remained untouched). This air and moisture tolerant reaction provides silyl-protected alcohols in a straightforward reduction-protection protocol as bulky hydrosilanes were used (HSiMe
2Ph, HSiEt
3, HSiMe
2Ph and HSitBuMe
2).
Scheme 1. ReI(O)2(PPh3)2 (C1) catalyzed hydrosilylation of carbonyl compounds with related mechanism studies. (a) Scope of the reaction; (b) Proposed [2 + 2] addition mechanistic pathway; (c) Proposed ionic outer-sphere mechanistic pathway.
A detailed mechanism, supported by experimental evidence, was proposed by the same group [
21,
25] and confirmed by a computational study by Wu et al. in 2006 [
26] (
Scheme 1b). The first step involves the formal [2 + 2] addition of silane to the Re=O bond in
C1 to produce metal hydride
I-1, which was isolated and characterized by X-ray diffraction. Alkoxy-metal intermediate
I-3 is produced by the addition of rhenium hydride to the carbonyl group. Transfer of the silyl group to the alkoxy ligand, formally a
retro-[2 + 2] reaction, forms the silyl ether product and regenerates the dioxo catalyst
C1.
However, the computational study by Wei [
27] showed that the ionic outer-sphere mechanistic pathway (
Scheme 1c) is energetically more favorable than the [2 + 2] addition mechanism supported by Wu for the
C1. In this alternative mechanism, the activation of Si−H goes through η
1-bonding of silane to the metal center followed by a nucleophilic addition of the organic substrate on the Si center in
II-1 to cleave the Si−H bond forming the anionic rhenium hydride
II-2 and the activated organic substrate
II-3. Finally, hydride transfer from the rhenium metal
II-2 to the carbon atom of the organic substrate
II-3 produces the desired product and regenerates complex
C1.
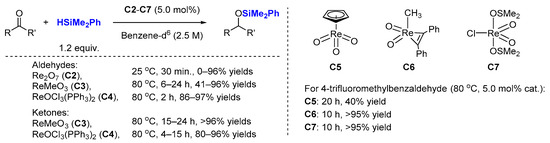
Intrigued by the work of Toste, Royo et al. explored in 2005 the catalytic activity of a family of oxo-rhenium complexes (
C2–
C7,
Scheme 2) [
28]. All these complexes showed activity in the reduction of aliphatic and aromatic aldehydes (six examples) and ketones (four examples) with dimethylphenylsilane in C
6D
6 solution (
Scheme 2), demonstrating the general ability of oxo-rhenium complexes to promote hydrosilylation. Notably,
C2 catalyzes the hydrosilylation of aldehydes at room temperature, within 30 min, affording the corresponding silyl ethers in a good yield, but is ineffective as a ketone hydrosilylation catalyst (even at 80 °C).
C5–
C7 (5.0 mol%) are active for the hydrosilylation of 4-trifluoromethylbenzaldehyde at 80 °C, giving 40% yield (
C5, 20 h), >95% yield (
C6, 10 h) and >95% yield (
C7, 10 h), respectively.
Scheme 2. Reduction of carbonyl groups by high-valent rhenium oxides C2–C7.
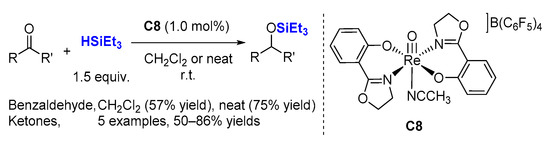
In the same year, Abu-Omar and co-workers reported a new system for the hydrosilylation of aldehydes and ketones using a mono-oxorhenium(V) catalyst (
C8) containing two oxazoline ligands (
Scheme 3) [
29,
30]. The reaction proceeds efficiently under ambient temperatures with low catalyst loading (0.1 mol%), 1.5 equiv. HSiEt
3 using CH
2Cl
2 as a solvent or without a solvent. Interestingly, the catalyst preserves its activity after being recycled (for 2-butanone, 80% NMR yield (1st cycle) and 50% NMR yield (2nd cycle)).
Scheme 3. Reduction of carbonyl groups by oxazoline containing rhenium oxide C8.
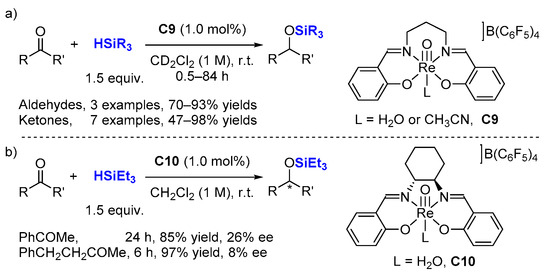
Then, Abu-Omar and co-workers prepared a number of cationic oxorhenium(V) salen based complexes such as [Re(O)(salpn)(Solv)][B(C
6F
5)
4]
C9 [
31,
32] in 2006 (
Scheme 4a) and [ReO(saldach)(H
2O)][B(C
6F
5)
4]
C10 [
33] in 2008 incorporating a chiral environment, (
Scheme 4b).
C9 and
C10 serve as good catalysts for hydrosilylation of carbonyl compounds (1.0 mol% cat. loading, 1.5 equiv. silane, room temperature (r.t.), although asymmetric versions (
C10) of these reactions afford poor enantioselectivity, even in the presence of bulky silane such as HSitBuMe
2.
Scheme 4. Hydrosilylation of carbonyl compounds with cationic oxorhenium(V) non chiral salen (a) and chiral salen (b) based complexes.
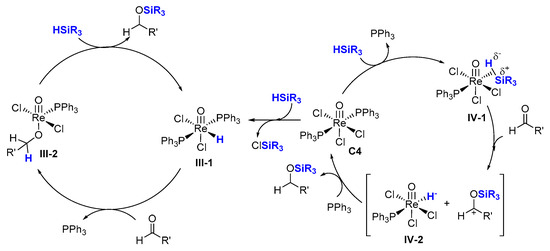
The mechanism of action of these high-valent mono-oxo-rhenium(V) catalysts was investigated, providing new insights [
34] including via a theoretical study [
35,
36,
37]. The addition of silane across the Re-O multiple bond was not observed in mono-oxo-rhenium(V) complex Re(O)Cl
3(PPh
3)
2 (
C4) catalyzing the hydrosilylation of carbonyls. Furthermore, they observed that silanes slowly reacted with
C4 to give the isolated hydride intermediate Re(O)Cl
2H(PPh
3)
2 (
III-1). However, this isolated hydride intermediate is not sufficiently reactive to account for the catalytic turnover, as only 20% yield of the silyl ether product is observed (
Scheme 5 left). These observations lead the authors to propose that the reaction pathway involves the formation of a η
2–silane complex (
IV-1), followed by the heterolytic cleavage of the Si-H bond at the rhenium center after elimination of one PPh
3 ligand (
Scheme 5, right). This ionic mechanism involves the sequential transfer of a silyl ion (R
3Si
+) and then a hydride to a polar double bond to furnish the reduction reactions. In this proposed catalytic cycle, neither the coordination of unsaturated substrates to the metal center nor its insertion into the M-H bond is involved.
Scheme 5. The σ-bond metathesis pathway involving the isolated hydride intermediate Re(O)Cl2H(PPh3)2 (left). The pathway involving a η2–silane complex (right).
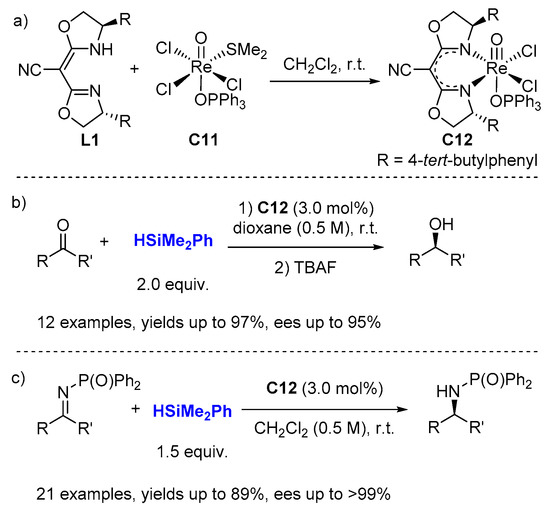
The enantioselective reduction of prochiral ketones [
38] and imines [
39] with 2 equiv. HSiMe
2Ph was reported by Toste (
Scheme 6) using chiral (CN-box)Re(V)–oxo complexes (3.0 mol%, CN-Box = cyanobis(oxazoline)). These reduction reactions proceed under an ambient air atmosphere with a highly functional group tolerant with ees of up to >99%. The (CNbox)Re(V)–oxo complexes (
C12) can be prepared by simply stirring
L1 with Re(O)Cl
3(OPPh
3)(SMe
2) (
C11) in CH
2Cl
2 at r.t. and isolated as a green stable solid (
Scheme 6a) or generated directly in situ.
Scheme 6. Enantioselective reduction of ketones (b) and imines (c) catalyzed by (CN-Box)Re(V)–oxo complexes C12 (a).
Similarly, a theoretical investigation of the mechanism for the high-valent mono-oxo-rhenium(V) hydride Re(O)HCl
2(PPh
3)
2 (
III-1) catalyzed hydrosilylation of C=N functionalities was performed by Wei and co-workers [
40]. These results suggest that an ionic S
N2-Si outer-sphere pathway proceeding via the heterolytic cleavage of the Si-H bond competes with the hydride pathway featuring the C=N bond inserted into the Re-H bond.
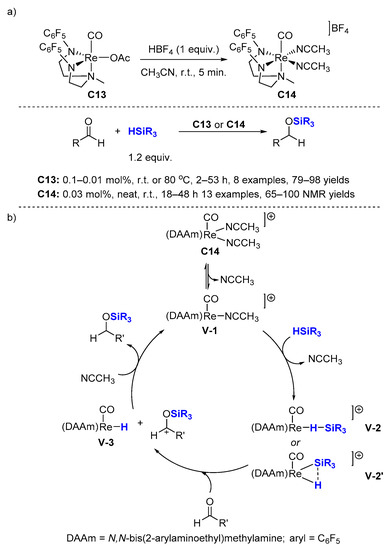
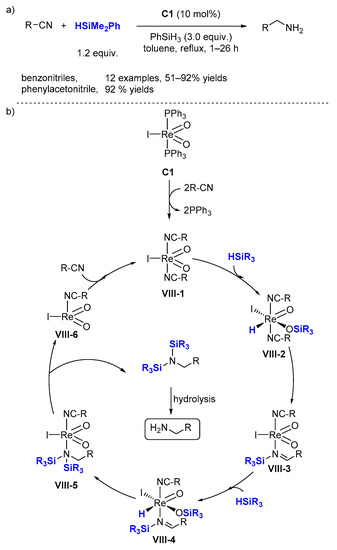
Besides these oxorhenium complexes, a series of novel low-valent Re(III) complexes were synthesized by Ison et al., such as
C13 [
41] in 2012 and
C14 [
42] in 2017 (
Scheme 7a).
C14 (0.03 mol%, r.t.) is more reactive than
C13 (0.1 mol%, 80 °C) for the hydrosilylation of aldehydes [
42]. Excellent NMR yields (65–100%, 13 examples) were achieved at ambient temperature under neat conditions using dimethylphenylsilane with
C14. The reaction affords TONs of up to 9200 and a TOF of up to 126 h
−1 (
Scheme 7a).
Scheme 7. Cationic rhenium complex catalyzed hydrosilylation of aldehydes. (a) Scope of the reaction; (b) Proposed mechanism.
Based on kinetic and mechanistic studies, an ionic outer-sphere mechanism for the catalytic hydrosilylation of benzaldehyde by
C14 has been proposed (
Scheme 7b). First, silane is activated through η
1 (
V-2) or η
2 (
V-2′) coordination to the rhenium center. Then, the aldehyde substrate attacks the Si center in (
V-2) or (
V-2′) to prompt the heterolytic cleavage of the Si-H bond yielding an ion pair, the hydride complex
V-3 and a silylated aldehyde ion. Finally, hydride transfer from rhenium to the carbonyl carbon of the activated substrate completes the catalytic cycle [
43].
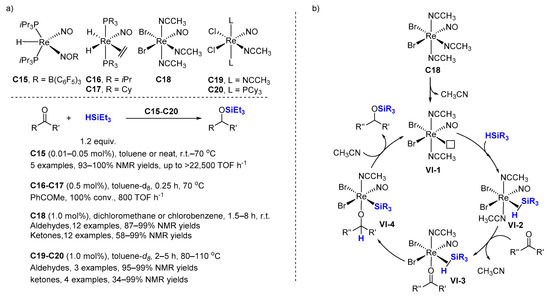
Berke and co-workers reported several easily available nitrosyl-rhenium complexes, for example, Re(H)(P
iPr
3)
2(NO)(NOB(C
6F
5)
3) (
C15) [
44] in 2005, Re(H)
2(η
2-C
2H
4)
3(NO)(PR
3)
2 (
C16, R =
iPr,
C17, R = Cy) [
45] in 2008, Re(CH
3CN)
3Br
2(NO) (
C18) [
46] in 2009, Re(CH
3CN)
3Cl
2(NO) (
C19) [
47] and Re(CH
3CN)Cl
2(NO)(PCy
3)
2 (
C20) [
47] in 2011. Those nitrosyl rhenium complexes were employed in the catalytic hydrosilylation of a variety of carbonyl compounds (
Scheme 8a). Hydrosilylations of carbonyl compounds were carried out with the catalyst
C15, HSiR
3 (1 equiv., R = Et or Ph) from r.t. to 70 °C either in toluene or under solvent-free conditions. TONs up to 9000 and TOFs up to 22,500 h
−1 were observed.
C16 and
C17 exhibited similar activity in hydrosilylation of acetophenone (0.5 mol% cat. loading at 70 °C in toluene-
d8, giving full conv. and TOF up to 800 h
−1).
Scheme 8. Rhenium oxides catalyzed hydrosilylation of carbonyl compounds. (a) Scope of the reaction; (b) Proposed mechanism.
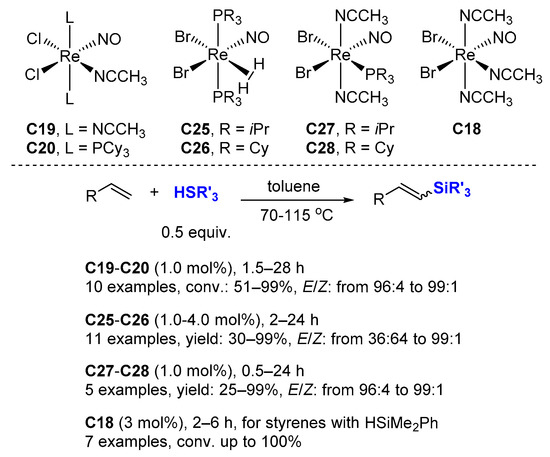
For C18, chlorobenzene was found to be superior to all the other solvents used. Various aliphatic and aromatic silanes were tested. Excellent yields were achieved at r.t. in dichloromethane using triethylsilane, the reaction affording TOF values of up to 495 h−1.
Phosphine-free complex C19 proved to be less effective than the bisphosphine derivative C20, as the reaction of benzophenone and Et3SiH (1.12 equiv.) with 1.0 mol% of C19 was carried out at 80 °C, a conversion of less than 10% was achieved within 4 h, while in the same conditions, C20 gave a 99% yield.
A possible mechanism for the hydrosilylation of ketones catalyzed by
C18 was proposed (
Scheme 8b). The initial step consists of the dissociation of a CH
3CN ligand to generate the pre-catalyst
VI-1 followed by the coordination of the silane to the rhenium center to form a η
2–silane complex
VI-2. Displacement of a second CH
3CN ligand by the ketone at the rhenium center yields complex
VI-3. The silyl-oxy complex
VI-4 could be obtained either from the transfer of a hydride from the silane to the ketone atom or from the oxidative addition of the silane followed by a β-hydride transfer process. Finally, the reductive elimination step produces the siloxy product and regenerates the pre-catalyst
VI-1.
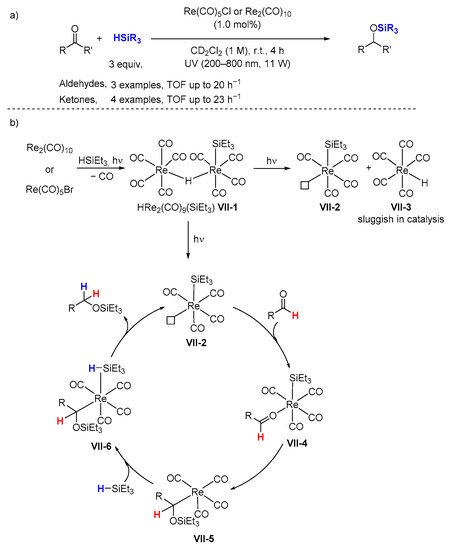
In 2012, carbonyl rhenium(I) complexes, namely Re(CO)
5Cl (
C21) and Re
2(CO)
10 (
C22), have been found by Fan and co-workers [
48] to be effective catalysts for the hydrosilylation of carbonyl substrates with various silanes and with TOF of 20−25 h
−1 for aldehydes (
Scheme 9a). In this methodology, 1.0 mol% catalyst and a Et
3SiH:carbonyl ratio of 3:1 were used. When different silanes such as Ph
2SiH
2 and Ph
3SiH were used, a decrease in the corresponding silyl ether yield was observed.
Scheme 9. Hydrosilylation of carbonyls via Re(CO)5Cl photolysis (a) and proposed mechanism (b).
A detailed mechanism for the hydrosilylation of carbonyl compounds was proposed by the same author to account for the experimental observations (
Scheme 9b). Upon photolysis of Et
3SiH with Re(CO)
5Cl or Re
2(CO)
10, a dimeric rhenium carbonyl species (
VII-1) with a bridging hydride was identified in the mixture. When
VII-1 was isolated and tested for aldehyde hydrosilylation, the silyl ether was generated about 2–3 times faster in comparison to Re(CO)
5Cl.
Upon photolysis, the dimer complex VII-1 dissociates to afford Et3SiRe(CO)4 (VII-2) and HRe(CO)5 (VII-3) which was found to be inactive in catalysis. Then, the coordination of the carbonyl substrate to the vacant site of VII-2 facilitates the silyl ligand shift onto the oxygen atom and forms an alkyl ligand bound to the Re center VII-5. Another silane coordinates to the Re center in VII-5 via a η2-silyl complex or a σ-silyl (σH) complex. Migration of a H atom from the silane to the alkyl group yields the silyl ether product and regenerates the catalyst VII-2. When either the carbonyl or silane has been depleted, the Et3SiRe(CO)4 complex(VII-2) coordinates back to HRe(CO)5 (VII-3) and becomes part of the resting state VII-1.
3. Hydrosilylation of C=C Bonds
3.1. Hydrosilylation of Alkenes
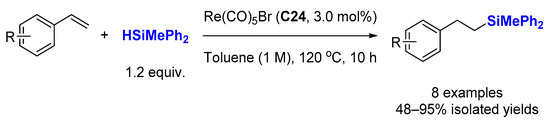
Despite the importance of the hydrosilylation of alkenes for the production of silicones, only limited examples have been reported with rhenium catalysts in contrast to the hydro-elementation of polar bonds. In 2006, the group of Hua reported the addition of hydrosilanes to styrenes catalyzed by low-valent rhenium complexes, such as Re(CO)
5Br (
C24), to selectively afford anti-Markovnikov adducts in good to high yields (
Scheme 15) [
58]. For the hydrosilylation of styrenes, 1.2 equiv. of HSiMePh
2 was employed at 120 °C in toluene (1 M) for 10 h. It is worth mentioning that other rhenium complexes, such as Re(CO)
5Cl (
C21) and Re
2(CO)
10 (
C22), also showed catalytic activity for the hydrosilylation of styrenes, although less actively than
C24, to give silylated alkanes in moderate yields as well as a slightly decreased selectivity. However, CpRe(CO)
3 (
C25) and NH
4ReO
4 (
C26) did not show any catalytic activity at all in such a transformation. Aliphatic alkenes, such as 1-octene and methyl methacrylate, were reduced to their corresponding adducts in poor yields (30% and 28%, respectively).
Scheme 15. Re(CO)5Br-catalyzed hydrosilylation of styrenes.
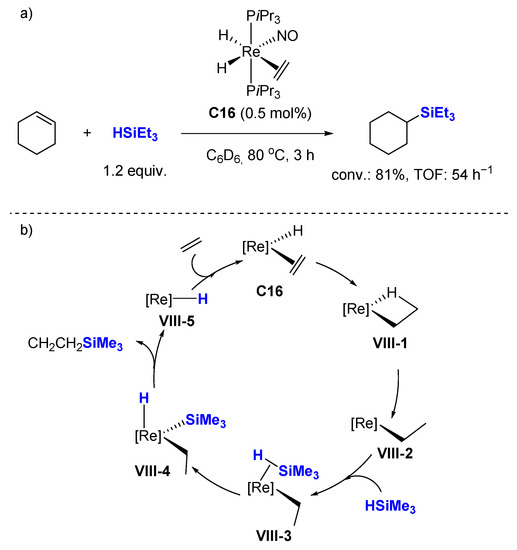
Interestingly, rhenium mono-nitrosyl complex
C16 developed by Berke et al., can also promote the catalytic hydrosilylation of cyclohexene [
45] (
Scheme 16a) under similar conditions that those employed for ketones (0.5 mol% cat., 80 °C, 3 h). This process was investigated theoretically by the group of Li with ethylene and trimethylsilane as model reactants (
Scheme 16b) [
59]. The proposed catalytic cycle starts with the insertion of the ethylene ligand into the Re-H bond, followed by the cleavage of the agostic interaction involved in
VIII-1 to afford
VIII-2. Then, a silane molecule coordinates to the Re center to give a σ complex
VIII-3. The next step involves an oxidative addition process to afford a seven coordinate complex
VIII-4. Last step consists of a reductive elimination process to form the product. The catalytic cycle completes by coordination of an ethylene to
VIII-5.
Scheme 16. Rhenium mono-nitrosyl complex catalyzed hydrosilylation of alkenes (a) and proposed mechanism (b).
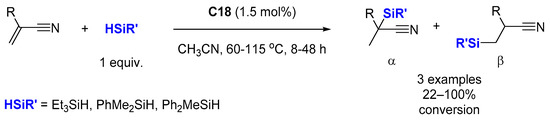
In 2014, the same group reported a highly selective hydrosilylation of acrylonitrile and its derivatives catalyzed by
C18 (1.5 mol%) by reaction with alkyl or aryl silanes (1 equiv.) at 60–115 °C in 8–48 h (
Scheme 17) [
60]. The regioselectivity of all the reactions were found to be around α:β = 3:1. Improved regioselectivity was obtained with a high ratio of acrylonitrile:silane.
Scheme 17. Re-catalyzed hydrosilylation of nitrile substituted olefins.
3.2. Dehydrogenative Silylation of Alkenes
Dehydrogenative silylation is often considered an undesired side reaction in the hydrosilylation of alkenes, but the selective production of vinylsilanes is also a valuable transformation. In this context; Berke et al. reported several rhenium mono-nitrosyl complexes (C18 [60], C19–C20 [47], C25–C26 [61], C27–C28 [62]) catalyzing the dehydrogenative silylation of alkenes with high chemoselectivity to produce silyl alkenes, along with the corresponding alkanes, with 1.0–4.0 mol% cat., at 70–110 °C in toluene-d8 (Scheme 18). Hydrosilylation products appear only rarely in very minor amounts. The complexes C19–C20 and C27–C28 gave better E/Z selectivity (96:4→99:1). Noticeably, ethylene can be transformed quantitatively into the corresponding dehydrogenative silylation product with C27–C28 as catalysts with a ratio dehydro/hydro silylation products of 79:21 and 68:32, respectively. C18 catalyzes the dehydrogenative coupling of styrene derivatives with HSiMe2Ph leading to dehydro/hydro silylation products in ratio of about 85/15.
Scheme 18. Rhenium mono-nitrosyl complexes catalyzed dehydrogenative silylation of alkenes.