 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Juan Antonio Rosado | + 3205 word(s) | 3205 | 2021-05-13 04:49:02 | | | |
| 2 | Vivi Li | Meta information modification | 3205 | 2021-05-19 10:49:31 | | |
Video Upload Options
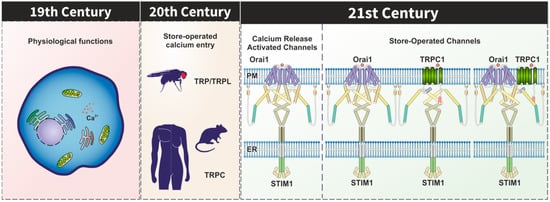
Transient receptor potential (TRP) proteins form non-selective Ca2+ permeable channels that contribute to the modulation of a number of physiological functions in a variety of cell types. Since the identification of TRP proteins in Drosophila, it is well known that these channels are activated by stimuli that induce PIP2 hydrolysis. The canonical TRP (TRPC) channels have long been suggested to be constituents of the store-operated Ca2+ (SOC) channels; however, none of the TRPC channels generate Ca2+ currents that resemble ICRAC. STIM1 and Orai1 have been identified as the components of the Ca2+ release-activated Ca2+ (CRAC) channels and there is a body of evidence supporting that STIM1 is able to gate Orai1 and TRPC1 in order to mediate non-selective cation currents named ISOC. STIM1 has been found to interact to and activate Orai1 and TRPC1 by different mechanisms and the involvement of TRPC1 in store-operated Ca2+ entry requires both STIM1 and Orai1. In addition to the participation of TRPC1 in the ISOC currents, TRPC1 and other TRPC proteins might play a relevant role modulating Orai1 channel function.
1. Introduction
| Orai1 Channels | Ora1-TRPC Channels | References | |
|---|---|---|---|
| Current Voltage (I–V) profile | Inwardly rectifying | Inwardly rectifying | [5][6][7] |
| Positive reversal potential ~ + 50 mV | Positive reversal potential 0 to ~ + 10 mV |
||
| Permeability and Selectivity | Ca2+ | K+, Na+, Cs+, Ca2+ and Ba2+ | [4][8] |
| Low to Cs3+ | |||
| Conduct Na+, Li+ and K+ in DVF solutions | |||
| Activation | Store depletion via STIM1 SOAR region | Store depletion via STIM1 SOAR and polibasic C-terminus regions | [9][10] |
| Endogenous current size | 0.1–0.2 pA/pF at −100 mV | [11] | |
| Fast Inactivation | Ca2+ | n/d | [12][13] |
| STIM1 CMD | |||
| Orai1 68–91 aa | |||
| Orai1 137–173 aa | |||
| Slow inactivation | Mitochondria | n/d | [14][15][16] |
| STIM1 390–391 aa | |||
| SARAF | |||
| Inhibitors | 2-APB (30–50 µM) | n/d | [17][18][19][20][21][22] |
| La3+ and Gd3+ (100 µM) | |||
| Low pH = 6.7 | |||
| Synta 66 | |||
| GSK-7975A GSK-5503A |
|||
| AnCOA4 (~5 µM) |
2. TRPC Channels in the STIM1–Orai1 Scenario
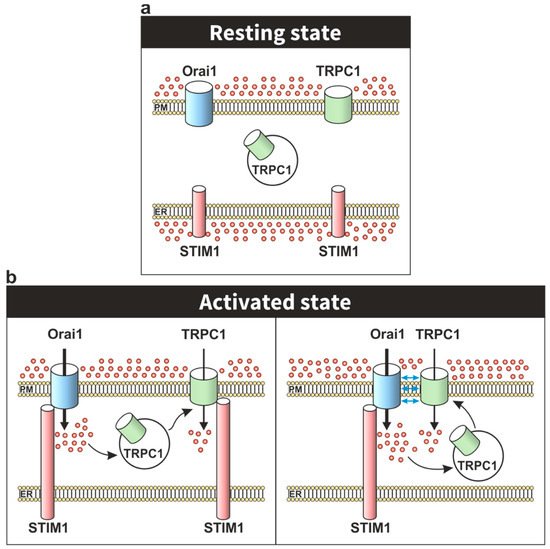
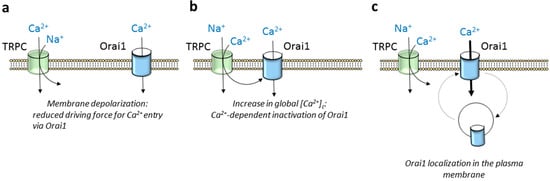
3. Modulation of Orai1 Function by TRPC Channels
References
- Ringer, S. A further Contribution regarding the influence of the different Constituents of the Blood on the Contraction of the Heart. J. Physiol. 1883, 4, 29–42.
- Putney, J.W., Jr. A model for receptor-regulated calcium entry. Cell Calcium 1986, 7, 1–12.
- Hoth, M.; Penner, R. Depletion of intracellular calcium stores activates a calcium current in mast cells. Nature 1992, 355, 353–356.
- Parekh, A.B.; Putney, J.W., Jr. Store-operated calcium channels. Physiol. Rev. 2005, 85, 757–810.
- Feske, S.; Gwack, Y.; Prakriya, M.; Srikanth, S.; Puppel, S.H.; Tanasa, B.; Hogan, P.G.; Lewis, R.S.; Daly, M.; Rao, A. A mutation in Orai1 causes immune deficiency by abrogating CRAC channel function. Nature 2006, 441, 179–185.
- Penner, R.; Matthews, G.; Neher, E. Regulation of calcium influx by second messengers in rat mast cells. Nature 1988, 334, 499–504.
- Kim, M.S.; Zeng, W.; Yuan, J.P.; Shin, D.M.; Worley, P.F.; Muallem, S. Native Store-operated Ca2+ Influx Requires the Channel Function of Orai1 and TRPC1. J. Biol. Chem. 2009, 284, 9733–9741.
- Parekh, A.B.; Penner, R. Store depletion and calcium influx. Physiol. Rev. 1997, 77, 901–930.
- Yuan, J.P.; Zeng, W.; Dorwart, M.R.; Choi, Y.J.; Worley, P.F.; Muallem, S. SOAR and the polybasic STIM1 domains gate and regulate Orai channels. Nat. Cell Biol. 2009, 11, 337–343.
- Zeng, W.; Yuan, J.P.; Kim, M.S.; Choi, Y.J.; Huang, G.N.; Worley, P.F.; Muallem, S. STIM1 gates TRPC channels, but not Orai1, by electrostatic interaction. Mol. Cell 2008, 32, 439–448.
- Zhang, X.; Gueguinou, M.; Trebak, M. Store-Independent Orai Channels Regulated by STIM. In Calcium Entry Channels in Non-Excitable Cells; Kozak, J.A., Putney, J.W., Jr., Eds.; Taylor and Francis Group: Boca Raton, FL, USA, 2018; pp. 197–214.
- Derler, I.; Fahrner, M.; Muik, M.; Lackner, B.; Schindl, R.; Groschner, K.; Romanin, C. A Ca2+release-activated Ca2+ (CRAC) modulatory domain (CMD) within STIM1 mediates fast Ca2+-dependent inactivation of ORAI1 channels. J. Biol. Chem. 2009, 284, 24933–24938.
- Lis, A.; Peinelt, C.; Beck, A.; Parvez, S.; Monteilh-Zoller, M.; Fleig, A.; Penner, R. CRACM1, CRACM2, and CRACM3 are store-operated Ca2+ channels with distinct functional properties. Curr. Biol. 2007, 17, 794–800.
- Albarran, L.; Lopez, J.J.; Gomez, L.J.; Salido, G.M.; Rosado, J.A. SARAF modulates TRPC1, but not TRPC6, channel function in a STIM1-independent manner. Biochem. J. 2016, 473, 3581–3595.
- Jha, A.; Ahuja, M.; Maleth, J.; Moreno, C.M.; Yuan, J.P.; Kim, M.S.; Muallem, S. The STIM1 CTID domain determines access of SARAF to SOAR to regulate Orai1 channel function. J. Cell Biol. 2013, 202, 71–79.
- Palty, R.; Raveh, A.; Kaminsky, I.; Meller, R.; Reuveny, E. SARAF inactivates the store operated calcium entry machinery to prevent excess calcium refilling. Cell 2012, 149, 425–438.
- Iwasaki, H.; Mori, Y.; Hara, Y.; Uchida, K.; Zhou, H.; Mikoshiba, K. 2-Aminoethoxydiphenyl borate (2-APB) inhibits capacitative calcium entry independently of the function of inositol 1,4,5-trisphosphate receptors. Recept. Channels 2001, 7, 429–439.
- Schindl, R.; Bergsmann, J.; Frischauf, I.; Derler, I.; Fahrner, M.; Muik, M.; Fritsch, R.; Groschner, K.; Romanin, C. 2-aminoethoxydiphenyl borate alters selectivity of Orai3 channels by increasing their pore size. J. Biol. Chem. 2008, 283, 20261–20267.
- Beech, D.J. Orai1 calcium channels in the vasculature. Pflug. Arch. 2012, 463, 635–647.
- Scrimgeour, N.; Litjens, T.; Ma, L.; Barritt, G.J.; Rychkov, G.Y. Properties of Orai1 mediated store-operated current depend on the expression levels of STIM1 and Orai1 proteins. J. Physiol. 2009, 587, 2903–2918.
- Derler, I.; Schindl, R.; Fritsch, R.; Heftberger, P.; Riedl, M.C.; Begg, M.; House, D.; Romanin, C. The action of selective CRAC channel blockers is affected by the Orai pore geometry. Cell Calcium 2013, 53, 139–151.
- Sadaghiani, A.M.; Lee, S.M.; Odegaard, J.I.; Leveson-Gower, D.B.; McPherson, O.M.; Novick, P.; Kim, M.R.; Koehler, A.N.; Negrin, R.; Dolmetsch, R.E.; et al. Identification of Orai1 channel inhibitors by using minimal functional domains to screen small molecule microarrays. Chem. Biol. 2014, 21, 1278–1292.
- Cosens, D.J.; Manning, A. Abnormal electroretinogram from a Drosophila mutant. Nature 1969, 224, 285–287.
- Minke, B.; Wu, C.; Pak, W.L. Induction of photoreceptor voltage noise in the dark in Drosophila mutant. Nature 1975, 258, 84–87.
- Hardie, R.C. Projection and connectivity of sex-specific photoreceptors in the compound eye of the male housefly (Musca domestica). Cell Tissue Res. 1983, 233, 1–21.
- Wes, P.D.; Chevesich, J.; Jeromin, A.; Rosenberg, C.; Stetten, G.; Montell, C. TRPC1, a human homolog of a Drosophila store-operated channel. Proc. Natl. Acad. Sci. USA 1995, 92, 9652–9656.
- Zhu, X.; Chu, P.B.; Peyton, M.; Birnbaumer, L. Molecular cloning of a widely expressed human homologue for the Drosophila trp gene. FEBS Lett. 1995, 373, 193–198.
- Venkatachalam, K.; Montell, C. TRP channels. Annu. Rev. Biochem. 2007, 76, 387–417.
- Montell, C. The TRP superfamily of cation channels. Sci. Singal. 2005, 2005, re3.
- Hellmich, U.A.; Gaudet, R. Structural biology of TRP channels. Handb. Exp. Pharmacol. 2014, 223, 963–990.
- Schaefer, M. Homo-and heteromeric assembly of TRP channel subunits. Pflug. Arch. 2005, 451, 35–42.
- Gregorio-Teruel, L.; Valente, P.; Gonzalez-Ros, J.M.; Fernandez-Ballester, G.; Ferrer-Montiel, A. Mutation of I696 and W697 in the TRP box of vanilloid receptor subtype I modulates allosteric channel activation. J. Gen. Physiol. 2014, 143, 361–375.
- Baez-Nieto, D.; Castillo, J.P.; Dragicevic, C.; Alvarez, O.; Latorre, R. Thermo-TRP channels: Biophysics of polymodal receptors. Adv. Exp. Med. Biol. 2011, 704, 469–490.
- Lee, K.P.; Choi, S.; Hong, J.H.; Ahuja, M.; Graham, S.; Ma, R.; So, I.; Shin, D.M.; Muallem, S.; Yuan, J.P. Molecular determinants mediating gating of Transient Receptor Potential Canonical (TRPC) channels by stromal interaction molecule 1 (STIM1). J. Biol. Chem. 2014, 289, 6372–6382.
- Wedel, B.J.; Vazquez, G.; McKay, R.R.; St, J.B.G.; Putney, J.W., Jr. A calmodulin/inositol 1,4,5-trisphosphate (IP3) receptor-binding region targets TRPC3 to the plasma membrane in a calmodulin/IP3 receptor-independent process. J. Biol. Chem. 2003, 278, 25758–25765.
- Dionisio, N.; Albarran, L.; Berna-Erro, A.; Hernandez-Cruz, J.M.; Salido, G.M.; Rosado, J.A. Functional role of the calmodulin-and inositol 1,4,5-trisphosphate receptor-binding (CIRB) site of TRPC6 in human platelet activation. Cell. Signal. 2011, 23, 1850–1856.
- Talavera, K.; Nilius, B. Electrophysiological Methods for the Study of TRP Channels. In TRP Channels; Zhu, M.X., Ed.; Taylor and Francis Group: Boca Raton, FL, USA, 2011.
- Yeromin, A.V.; Zhang, S.L.; Jiang, W.; Yu, Y.; Safrina, O.; Cahalan, M.D. Molecular identification of the CRAC channel by altered ion selectivity in a mutant of Orai. Nature 2006, 443, 226–229.
- Luik, R.M.; Wu, M.M.; Buchanan, J.; Lewis, R.S. The elementary unit of store-operated Ca2+ entry: Local activation of CRAC channels by STIM1 at ER-plasma membrane junctions. J. Cell Biol. 2006, 174, 815–825.
- Vig, M.; Beck, A.; Billingsley, J.M.; Lis, A.; Parvez, S.; Peinelt, C.; Koomoa, D.L.; Soboloff, J.; Gill, D.L.; Fleig, A.; et al. CRACM1 multimers form the ion-selective pore of the CRAC channel. Curr. Biol. 2006, 16, 2073–2079.
- Prakriya, M.; Feske, S.; Gwack, Y.; Srikanth, S.; Rao, A.; Hogan, P.G. Orai1 is an essential pore subunit of the CRAC channel. Nature 2006, 443, 230–233.
- Darbellay, B.; Arnaudeau, S.; Bader, C.R.; Konig, S.; Bernheim, L. STIM1L is a new actin-binding splice variant involved in fast repetitive Ca2+ release. J. Cell Biol. 2011, 194, 335–346.
- Desai, P.N.; Zhang, X.; Wu, S.; Janoshazi, A.; Bolimuntha, S.; Putney, J.W.; Trebak, M. Multiple types of calcium channels arising from alternative translation initiation of the Orai1 message. Sci. Signal. 2015, 8, ra74.
- Fukushima, M.; Tomita, T.; Janoshazi, A.; Putney, J.W. Alternative translation initiation gives rise to two isoforms of Orai1 with distinct plasma membrane mobilities. J. Cell Sci. 2012, 125, 4354–4361.
- Stathopulos, P.B.; Zheng, L.; Ikura, M. Stromal interaction molecule (STIM) 1 and STIM2 calcium sensing regions exhibit distinct unfolding and oligomerization kinetics. J. Biol. Chem. 2009, 284, 728–732.
- Miederer, A.M.; Alansary, D.; Schwar, G.; Lee, P.H.; Jung, M.; Helms, V.; Niemeyer, B.A. A STIM2 splice variant negatively regulates store-operated calcium entry. Nat. Commun. 2015, 6, 6899.
- Rana, A.; Yen, M.; Sadaghiani, A.M.; Malmersjo, S.; Park, C.Y.; Dolmetsch, R.E.; Lewis, R.S. Alternative splicing converts STIM2 from an activator to an inhibitor of store-operated calcium channels. J. Cell Biol. 2015, 209, 653–669.
- Vaeth, M.; Yang, J.; Yamashita, M.; Zee, I.; Eckstein, M.; Knosp, C.; Kaufmann, U.; Karoly Jani, P.; Lacruz, R.S.; Flockerzi, V.; et al. ORAI2 modulates store-operated calcium entry and T cell-mediated immunity. Nat. Commun. 2017, 8, 14714.
- Wang, J.; Xu, C.; Zheng, Q.; Yang, K.; Lai, N.; Wang, T.; Tang, H.; Lu, W. Orai1, 2, 3 and STIM1 promote store-operated calcium entry in pulmonary arterial smooth muscle cells. Cell Death Discov. 2017, 3, 17074.
- Wei, D.; Mei, Y.; Xia, J.; Hu, H. Orai1 and Orai3 Mediate Store-Operated Calcium Entry Contributing to Neuronal Excitability in Dorsal Root Ganglion Neurons. Front. Cell. Neurosci. 2017, 11, 400.
- Zitt, C.; Zobel, A.; Obukhov, A.G.; Harteneck, C.; Kalkbrenner, F.; Luckhoff, A.; Schultz, G. Cloning and functional expression of a human Ca2+-permeable cation channel activated by calcium store depletion. Neuron 1996, 16, 1189–1196.
- Zhu, X.; Jiang, M.; Peyton, M.; Boulay, G.; Hurst, R.; Stefani, E.; Birnbaumer, L. Trp, a novel mammalian gene family essential for agonist-activated capacitative Ca2+ entry. Cell 1996, 85, 661–671.
- Liu, X.; Wang, W.; Singh, B.B.; Lockwich, T.; Jadlowiec, J.; O’Connell, B.; Wellner, R.; Zhu, M.X.; Ambudkar, I.S. Trp1, a candidate protein for the store-operated Ca2+ influx mechanism in salivary gland cells. J. Biol. Chem. 2000, 275, 3403–3411.
- Brough, G.H.; Wu, S.; Cioffi, D.; Moore, T.M.; Li, M.; Dean, N.; Stevens, T. Contribution of endogenously expressed Trp1 to a Ca2+-selective, store-operated Ca2+ entry pathway. FASEB J. 2001, 15, 1727–1738.
- Rosado, J.A.; Brownlow, S.L.; Sage, S.O. Endogenously expressed Trp1 is involved in store-mediated Ca2+ entry by conformational coupling in human platelets. J. Biol. Chem. 2002, 277, 42157–42163.
- Zitt, C.; Obukhov, A.G.; Strubing, C.; Zobel, A.; Kalkbrenner, F.; Luckhoff, A.; Schultz, G. Expression of TRPC3 in Chinese hamster ovary cells results in calcium-activated cation currents not related to store depletion. J. Cell Biol. 1997, 138, 1333–1341.
- Trebak, M.; St, J.B.G.; McKay, R.R.; Birnbaumer, L.; Putney, J.W., Jr. Signaling mechanism for receptor-activated canonical transient receptor potential 3 (TRPC3) channels. J. Biol. Chem. 2003, 278, 16244–16252.
- Jardin, I.; Lopez, J.J.; Salido, G.M.; Rosado, J.A. Orai1 mediates the interaction between STIM1 and hTRPC1 and regulates the mode of activation of hTRPC1-forming Ca2+ channels. J. Biol. Chem. 2008, 283, 25296–25304.
- Ong, H.L.; Cheng, K.T.; Liu, X.; Bandyopadhyay, B.C.; Paria, B.C.; Soboloff, J.; Pani, B.; Gwack, Y.; Srikanth, S.; Singh, B.B.; et al. Dynamic assembly of TRPC1-STIM1-Orai1 ternary complex is involved in store-operated calcium influx. Evidence for similarities in store-operated and calcium release-activated calcium channel components. J. Biol. Chem. 2007, 282, 9105–9116.
- Cheng, K.T.; Liu, X.; Ong, H.L.; Ambudkar, I.S. Functional requirement for Orai1 in store-operated TRPC1-STIM1 channels. J. Biol. Chem. 2008, 283, 12935–12940.
- Lopez, J.J.; Salido, G.M.; Pariente, J.A.; Rosado, J.A. Interaction of STIM1 with endogenously expressed human canonical TRP1 upon depletion of intracellular Ca2+ stores. J. Biol. Chem. 2006, 281, 28254–28264.
- Yuan, J.P.; Zeng, W.; Huang, G.N.; Worley, P.F.; Muallem, S. STIM1 heteromultimerizes TRPC channels to determine their function as store-operated channels. Nat. Cell Biol. 2007, 9, 636–645.
- Jardin, I.; Salido, G.M.; Rosado, J.A. Role of lipid rafts in the interaction between hTRPC1, Orai1 and STIM1. Channels 2008, 2, 401–403.
- Liao, Y.; Plummer, N.W.; George, M.D.; Abramowitz, J.; Zhu, M.X.; Birnbaumer, L. A role for Orai in TRPC-mediated Ca2+ entry suggests that a TRPC:Orai complex may mediate store and receptor operated Ca2+ entry. Proc. Natl. Acad. Sci. USA 2009, 106, 3202–3206.
- Derler, I.; Plenk, P.; Fahrner, M.; Muik, M.; Jardin, I.; Schindl, R.; Gruber, H.J.; Groschner, K.; Romanin, C. The extended transmembrane Orai1 N-terminal (ETON) region combines binding interface and gate for Orai1 activation by STIM1. J. Biol. Chem. 2013, 288, 29025–29034.
- Stathopulos, P.B.; Schindl, R.; Fahrner, M.; Zheng, L.; Gasmi-Seabrook, G.M.; Muik, M.; Romanin, C.; Ikura, M. STIM1/Orai1 coiled-coil interplay in the regulation of store-operated calcium entry. Nat. Commun. 2013, 4, 2963.
- Fahrner, M.; Muik, M.; Schindl, R.; Butorac, C.; Stathopulos, P.; Zheng, L.; Jardin, I.; Ikura, M.; Romanin, C. A coiled-coil clamp controls both conformation and clustering of stromal interaction molecule 1 (STIM1). J. Biol. Chem. 2014, 289, 33231–33244.
- Jia, S.; Rodriguez, M.; Williams, A.G.; Yuan, J.P. Homer binds to Orai1 and TRPC channels in the neointima and regulates vascular smooth muscle cell migration and proliferation. Sci. Rep. 2017, 7, 5075.
- Cheng, K.T.; Liu, X.; Ong, H.L.; Swaim, W.; Ambudkar, I.S. Local Ca2+ entry via Orai1 regulates plasma membrane recruitment of TRPC1 and controls cytosolic Ca2+ signals required for specific cell functions. PLoS Biol. 2011, 9, e1001025.
- Ambudkar, I.S.; de Souza, L.B.; Ong, H.L. TRPC1, Orai1, and STIM1 in SOCE: Friends in tight spaces. Cell Calcium 2017, 63, 33–39.
- Ong, H.L.; Jang, S.I.; Ambudkar, I.S. Distinct contributions of Orai1 and TRPC1 to agonist-induced [Ca2+](i) signals determine specificity of Ca2+-dependent gene expression. PLoS ONE 2012, 7, e47146.
- Galan, C.; Zbidi, H.; Bartegi, A.; Salido, G.M.; Rosado, J.A. STIM1, Orai1 and hTRPC1 are important for thrombin- and ADP-induced aggregation in human platelets. Arch. Biochem. Biophys. 2009, 490, 137–144.
- Sabourin, J.; Le Gal, L.; Saurwein, L.; Haefliger, J.A.; Raddatz, E.; Allagnat, F. Store-operated Ca2+ Entry Mediated by Orai1 and TRPC1 Participates to Insulin Secretion in Rat beta-Cells. J. Biol. Chem. 2015, 290, 30530–30539.
- Schaar, A.; Sun, Y.; Sukumaran, P.; Rosenberger, T.A.; Krout, D.; Roemmich, J.N.; Brinbaumer, L.; Claycombe-Larson, K.; Singh, B.B. Ca2+ entry via TRPC1 is essential for cellular differentiation and modulates secretion via the SNARE complex. J. Cell Sci. 2019, 132.
- Perrouin-Verbe, M.A.; Schoentgen, N.; Talagas, M.; Garlantezec, R.; Uguen, A.; Doucet, L.; Rosec, S.; Marcorelles, P.; Potier-Cartereau, M.; Vandier, C.; et al. Overexpression of certain transient receptor potential and Orai channels in prostate cancer is associated with decreased risk of systemic recurrence after radical prostatectomy. Prostate 2019, 79, 1793–1804.
- Gutierrez, L.G.; Hernandez-Morales, M.; Nunez, L.; Villalobos, C. Inhibition of Polyamine Biosynthesis Reverses Ca2+ Channel Remodeling in Colon Cancer Cells. Cancers 2019, 11, 83.
- Gueguinou, M.; Harnois, T.; Crottes, D.; Uguen, A.; Deliot, N.; Gambade, A.; Chantome, A.; Haelters, J.P.; Jaffres, P.A.; Jourdan, M.L.; et al. SK3/TRPC1/Orai1 complex regulates SOCE-dependent colon cancer cell migration: A novel opportunity to modulate anti-EGFR mAb action by the alkyl-lipid Ohmline. Oncotarget 2016, 7, 36168–36184.
- Sabourin, J.; Boet, A.; Rucker-Martin, C.; Lambert, M.; Gomez, A.M.; Benitah, J.P.; Perros, F.; Humbert, M.; Antigny, F. Ca2+ handling remodeling and STIM1L/Orai1/TRPC1/TRPC4 upregulation in monocrotaline-induced right ventricular hypertrophy. J. Mol. Cell. Cardiol. 2018, 118, 208–224.
- Nunez, L.; Bird, G.S.; Hernando-Perez, E.; Perez-Riesgo, E.; Putney, J.W., Jr.; Villalobos, C. Store-operated Ca2+ entry and Ca2+ responses to hypothalamic releasing hormones in anterior pituitary cells from Orai1-/-and heptaTRPC knockout mice. Biochim. Biophys. Acta Mol. Cell Res. 2019, 1866, 1124–1136.
- Shi, J.; Miralles, F.; Kinet, J.P.; Birnbaumer, L.; Large, W.A.; Albert, A.P. Evidence that Orai1 does not contribute to store-operated TRPC1 channels in vascular smooth muscle cells. Channels 2017, 11, 329–339.
- Darbellay, B.; Arnaudeau, S.; Konig, S.; Jousset, H.; Bader, C.; Demaurex, N.; Bernheim, L. STIM1- and Orai1-dependent store-operated calcium entry regulates human myoblast differentiation. J. Biol. Chem. 2009, 284, 5370–5380.
- Antigny, F.; Koenig, S.; Bernheim, L.; Frieden, M. During post-natal human myogenesis, normal myotube size requires TRPC1-and TRPC4-mediated Ca2+ entry. J. Cell Sci. 2013, 126, 2525–2533.
- Antigny, F.; Sabourin, J.; Sauc, S.; Bernheim, L.; Koenig, S.; Frieden, M. TRPC1 and TRPC4 channels functionally interact with STIM1L to promote myogenesis and maintain fast repetitive Ca2+ release in human myotubes. Biochim. Biophys. Acta Mol. Cell Res. 2017, 1864, 806–813.
- Fatherazi, S.; Presland, R.B.; Belton, C.M.; Goodwin, P.; Al-Qutub, M.; Trbic, Z.; Macdonald, G.; Schubert, M.M.; Izutsu, K.T. Evidence that TRPC4 supports the calcium selective I(CRAC)-like current in human gingival keratinocytes. Pflug. Arch. 2007, 453, 879–889.
- Sundivakkam, P.C.; Freichel, M.; Singh, V.; Yuan, J.P.; Vogel, S.M.; Flockerzi, V.; Malik, A.B.; Tiruppathi, C. The Ca2+ sensor stromal interaction molecule 1 (STIM1) is necessary and sufficient for the store-operated Ca2+ entry function of transient receptor potential canonical (TRPC) 1 and 4 channels in endothelial cells. Mol. Pharmacol. 2012, 81, 510–526.
- Brechard, S.; Melchior, C.; Plancon, S.; Schenten, V.; Tschirhart, E.J. Store-operated Ca2+ channels formed by TRPC1, TRPC6 and Orai1 and non-store-operated channels formed by TRPC3 are involved in the regulation of NADPH oxidase in HL-60 granulocytes. Cell Calcium 2008, 44, 492–506.
- Jardin, I.; Redondo, P.C.; Salido, G.M.; Rosado, J.A. Phosphatidylinositol 4,5-bisphosphate enhances store-operated calcium entry through hTRPC6 channel in human platelets. Biochim. Biophys. Acta 2008, 1783, 84–97.
- Selli, C.; Erac, Y.; Kosova, B.; Tosun, M. Post-transcriptional silencing of TRPC1 ion channel gene by RNA interference upregulates TRPC6 expression and store-operated Ca2+ entry in A7r5 vascular smooth muscle cells. Vasc. Pharmacol. 2009, 51, 96–100.
- Jardin, I.; Gomez, L.J.; Salido, G.M.; Rosado, J.A. Dynamic interaction of hTRPC6 with the Orai1/STIM1 complex or hTRPC3 mediates its role in capacitative or non-capacitative Ca2+ entry pathways. Biochem. J. 2009, 420, 267–276.
- Gees, M.; Colsoul, B.; Nilius, B. The role of transient receptor potential cation channels in Ca2+ signaling. Cold Spring Harb. Perspect. Biol. 2010, 2, a003962.
- Zweifach, A.; Lewis, R.S. Rapid inactivation of depletion-activated calcium current (ICRAC) due to local calcium feedback. J. Gen. Physiol. 1995, 105, 209–226.
- Fierro, L.; Parekh, A.B. Fast calcium-dependent inactivation of calcium release-activated calcium current (CRAC) in RBL-1 cells. J. Membr. Biol. 1999, 168, 9–17.
- Zweifach, A.; Lewis, R.S. Slow calcium-dependent inactivation of depletion-activated calcium current. Store-dependent and -independent mechanisms. J. Biol. Chem. 1995, 270, 14445–14451.
- Villalobos, C.; Gutierrez, L.G.; Hernandez-Morales, M.; Del Bosque, D.; Nunez, L. Mitochondrial control of store-operated Ca2+ channels in cancer: Pharmacological implications. Pharmacol. Res. 2018, 135, 136–143.
- Launay, P.; Cheng, H.; Srivatsan, S.; Penner, R.; Fleig, A.; Kinet, J.P. TRPM4 regulates calcium oscillations after T cell activation. Science 2004, 306, 1374–1377.
- Avila-Medina, J.; Calderon-Sanchez, E.; Gonzalez-Rodriguez, P.; Monje-Quiroga, F.; Rosado, J.A.; Castellano, A.; Ordonez, A.; Smani, T. Orai1 and TRPC1 Proteins Co-localize with CaV1.2 Channels to Form a Signal Complex in Vascular Smooth Muscle Cells. J. Biol. Chem. 2016, 291, 21148–21159.
- Beck, A.; Gotz, V.; Qiao, S.; Weissgerber, P.; Flockerzi, V.; Freichel, M.; Boehm, U. Functional Characterization of Transient Receptor Potential (TRP) Channel C5 in Female Murine Gonadotropes. Endocrinology 2017, 158, 887–902.
- Schindl, R.; Fritsch, R.; Jardin, I.; Frischauf, I.; Kahr, H.; Muik, M.; Riedl, M.C.; Groschner, K.; Romanin, C. Canonical transient receptor potential (TRPC) 1 acts as a negative regulator for vanilloid TRPV6-mediated Ca2+ influx. J. Biol. Chem. 2012, 287, 35612–35620.
- Moore, T.M.; Brough, G.H.; Babal, P.; Kelly, J.J.; Li, M.; Stevens, T. Store-operated calcium entry promotes shape change in pulmonary endothelial cells expressing Trp1. Am. J. Physiol. 1998, 275, L574–L582.
- Jardin, I.; Diez-Bello, R.; Lopez, J.J.; Redondo, P.C.; Salido, G.M.; Smani, T.; Rosado, J.A. TRPC6 Channels Are Required for Proliferation, Migration and Invasion of Breast Cancer Cell Lines by Modulation of Orai1 and Orai3 Surface Exposure. Cancers 2018, 10, 331.
- Diez-Bello, R.; Jardin, I.; Lopez, J.J.; El Haouari, M.; Ortega-Vidal, J.; Altarejos, J.; Salido, G.M.; Salido, S.; Rosado, J.A. (-)Oleocanthal inhibits proliferation and migration by modulating Ca2+ entry through TRPC6 in breast cancer cells. Biochim. Biophys. Acta Mol. Cell Res. 2019, 1866, 474–485.

