 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Ruth Stuckey | + 7164 word(s) | 7164 | 2021-05-17 13:29:56 | | | |
| 2 | Conner Chen | Meta information modification | 7164 | 2021-05-25 09:34:15 | | |
Video Upload Options
Polycythaemia Vera (PV) is a BCR-ABL1-negative MPN characterized by the clonal proliferation of hematopoietic progenitor cells, resulting in an increased production of morphologically normal red blood cells, white blood cells and platelets, although erythrocytosis most often predominates. The disorder is most common in those aged 50–70 years, with an incidence that increases with older age. Patients diagnosed with PV have a shorter survival compared to the age-matched general population, with a median survival of 13.5 years, due to an increased predisposition for thrombosis as well as transformation to myelofibrosis (post-PV myelofibrosis, estimated to occur in 10% of patients with PV) or acute myeloid leukaemia (AML, estimated to occur in 15% of patients with PV).
1. Diagnosis of PV
Diagnosis of PV is made according to the 2016 WHO criteria and is based on the assessment of several clinical, haematological and molecular features, including serum haemoglobin level, BCR-ABL1-negativity, and JAK2 mutation status [1][2]. Here, we will focus on how the molecular testing of genetic alterations can inform a more precise diagnosis of PV and recommend readers to two comprehensive reviews for general guidelines on the diagnosis and management of patients with PV [3][4].
1.1. Driver Mutations for PV
The key driver gene in the BCR-ABL1-negative MPNs PV, ET and PMF is JAK2, encoding the tyrosine kinase Janus kinase 2, a critical mediator in erythropoiesis [5][6][7]. The point mutation c.1849G>T, affecting exon 14, causes a valine to phenylalanine substitution at position 617 in the JH2 domain.
The JAK2V617F variant is the most frequent mutation in BCR-ABL1-negative MPN, detected in 97% of PV [8], 50–60% of ET, and 55–60% of PMF cases [9]. Unlike the MPNs ET and PMF, mutations in MPL and CALR are not found in patients with PV. However, one study reported two JAK2V617F-negative PV patients who harboured the CALR type 1 (52-bp del) mutation in peripheral granulocytes at diagnosis [10]. Both patients had haemoglobin above the threshold for diagnosis (including according to the WHO’s 2016 revised criteria), moderately elevated platelet counts and normal leukocyte counts, and bone marrow biopsy showed hypercellularity. No mutation was found in JAK2 exons 12, 13, and 14 or in MPL exon 10 for either patient. Nevertheless, this is only an isolated report, and subsequent studies, including the retrospective molecular diagnosis of 524 JAK2V617F-negative patients with suspected MPN, have failed to identify CALR mutations in patients with PV [11].
If the molecular analysis of JAK2V617F is negative, analysis for mutations in exon 12, such as N542-E543del, E543-D544del and K539L, is indicated [12] since mutations in exon 12 of JAK2 are responsible for the majority of the remaining JAK2V617F-negative PV patients. These exon 12 mutations consist of deletions, insertions, duplications and substitutions of bases that affect the region linking the SH2 and JH2 domains and lead to the constitutive activation of the kinase [13]. Nevertheless, a study by Skoda and colleagues found that JAK2V617F and other exon 12 mutations can coexist in two separate clones in some patients [14].
Interestingly, some studies (including those performed using mouse models) have suggested that mutations in exon 12 of JAK2 may confer a different clinical phenotype to JAK2V617F. For example, patients with exon 12 mutations were younger and frequently presented with erythrocytosis [15][16][17]. Nevertheless, in a European collaborative study, 106 patients harbouring the most frequent exon 12 mutations were reported to have similar rates of survival and constitutive symptoms to patients with JAK2V617F [13].
1.2. Techniques for Detection of the JAK2V617F Variant
According to the European LeukemiaNet (ELN), the standardized technique for the detection of the JAK2V617F variant is based on allele-specific PCR, which also allows for the quantification of the allelic frequency [18][19]. The technique of digital PCR (dPCR) is increasingly being employed since it permits the absolute quantification of the JAK2V617F variant without the need for the prior use of a standard curve [20][21]. For the detection of other JAK2 variants, Sanger sequencing of genomic DNA is often employed, although it can miss mutations with allele frequencies less than 10–20% [12].
In recent years, more sensitive detection methods, such as sequencing of plasma RNA [22] and NGS [23][24], are being increasingly employed for the molecular diagnosis of patients with PV. NGS provides the advantage of potentially detecting all JAK2 variants, including rare and/or previously undescribed mutations [23].
The most recently developed technique for the genetic profiling of patients with MPN and other haematological malignancies is the isolation of cell-free circulating DNA (ccfDNA) from patient plasma (also commonly known as liquid biopsy) coupled with its analysis by NGS [25]. A recent study assessed the feasibility of mutational analysis by the NGS of 107 patients with MPN (including 33 patients with PV) comparing the use of ccfDNA from plasma versus genomic DNA isolated from granulocytes. The authors confirmed that the sensitivity and accuracy were similar for both types of genetic material tested, with a similar mutational profile identified. However, the median VAF detected was significantly higher in ccfDNA than in DNA extracted from granulocytes (29% versus 25%, respectively) [26].
Nevertheless, despite the use of more sensitive techniques, 2–3% of patients with PV are estimated to be JAK2 wild type. Approximately 2% of such JAK2-negative PV patients harbour a mutation in the gene SH2B3 (also known as LNK), which encodes an inhibitor of cytokine-dependent cell growth in hematopoietic cells. Thus, loss of function mutations in SH2B3 causes the sustained activation of JAK2 resulting in erythrocytosis [27][28]. Indeed, Lnk−/− mice models developed an MPN-like phenotype, with abnormal megakaryopoiesis and erythropoiesis, cytokine hypersensitivity, splenomegaly, fibrosis, and extramedullary haematopoiesis [29][30]. The search for mutations in SH2B3 in JAK2-negative patients suspected of having PV is recommended [31], particularly in the hotspot in exon 2, although mutations in SH2B3 have also been described in PV patients with concomitant JAK2V617F [32].
In summary, when considering the molecular analysis of patients with a suspicion of PV, it is important that (i) techniques with a sensitivity of at least 5% are employed to search for driver mutations, and (ii) such testing is not simply limited to the detection of the JAK2V617F variant or other exon 12 mutations. For JAK2V617F-negative PV patients, additional mutational screening is justified, first in other exons followed by the SH2B3 gene (Figure 1), despite the additional time and financial costs involved [31].
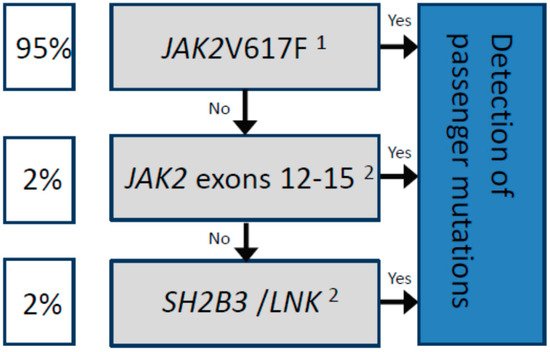
Figure 1. Sequential molecular analysis for the diagnosis of patients with polycythaemia vera.
Driver mutations are represented in the gray boxes. The estimated frequency among patients diagnosed with PV is shown in the white boxes. In all cases, the concomitant detection of passenger mutations by NGS is recommended (blue box) for a more accurate prognosis, for example, to predict transformation to MF or AML, as well as to detect mutations associated with increased thrombotic risk, such as TET2. 1The standardized technique according to the European LeukemiaNet (ELN) is based on allele-specific PCR, which allows for the detection of the JAK2V617F mutation as well as the quantification of the allelic frequency [18]. 2The use of next-generation sequencing (NGS) is highly recommended, or other molecular techniques with a sensitivity of at least 5%.
1.3. Biological Impact of JAK2 Driver Mutations
The JAK2 kinase interacts with the erythropoietin (Epo), thrombopoietin (Tpo), and granulocyte colony-stimulating factor (G-CSF) receptors to regulate the normal proliferation, differentiation, and survival of myeloid cells [33]. The JAK2V617F driver mutation and other gain-of-function JAK2 variants that cause the constitutive activation of the JAK2 kinase induce the proliferation of hematopoietic stem cells and provide them with a survival advantage [34][35][36]. The aberrant activation of JAK2 leads to signalling via the erythropoietin (Epo) receptor without cytokine stimulation [7][35][36] and the overactivation of several downstream signalling pathways, including JAK2/STAT5, RAS/MAPK/ERK and PI3K pathways [37]. In addition to STAT5, STAT3 is also activated in MPNs, with the phosphorylation of STAT3 and STAT5 shown to be uniformly increased in PV, in contrast to ET and PMF.
The JAK2V617F mutation has been shown to be clonal [7][35][36] and present in haematopoietic stem cells (HSCs) [37]. Several studies have demonstrated that the JAK2V617F mutant promotes genomic instability via two mechanisms: increasing levels of DNA damage and decreasing DNA damage-induced cell death. For example, aberrant JAK2/STAT5 and PI3K signalling lead to an accumulation of DNA damage via the production of high levels of reactive oxygen species and the stimulation of homologous recombination, leading to increased amounts of double-strand breaks [38][39][40][41] that in turn impede replication fork progression [42].
Indeed, Quelle et al. linked the misregulation of the JAK2 signalling pathway to the suppression of p53-mediated cell death in response to DNA damage in hematopoietic cells as early as 1998 [34]. JAK2/STAT5 activation promotes the G1-S phase transition of the cell cycle due to the increased degradation of p53 as a result of enhanced MDM2 translation [39][43]. Impairment of the intra-S checkpoint response in erythroblasts from PV but not ET patients was also shown to result in increased DNA damage due to attenuated p53 signalling [42]. Furthermore, JAK2/STAT5 pathway activation induces overexpression of the STAT5 target gene BCL2L1, which codes for the anti-apoptotic protein Bcl-xl [34][44], as well as inhibiting its deamidation [45] preventing apoptosis in response to the detection of DNA damage. Together, these mechanisms result in an accumulation of proliferating malignant cells with unresolved DNA damage.
Interestingly, treatment of mice engrafted with JAK2V617F-mutated bone marrow cells with the antioxidant N-acetylcysteine resulted in reduced levels of DNA damage and decreased splenomegaly [41], while the dietary intervention of patients with MPN, including N-acetylcysteine supplementation, led to a certain improvement in symptom burden [45][46]. These results suggest that the reduction of damaging ROS effects with antioxidants may potentially be a promising therapeutic approach.
The principal model of pathogenesis of PV and other MPN is based on genomic instability; however, increasing evidence is also supporting the consideration that the clonal evolution characteristic of MPN and other cancers may be triggered by chronic inflammation and an escape from anti-tumour immunological surveillance [47][48]. Indeed, the incidence of second cancer has been reported in up to 10% of all MPN patients [49][50], with a second solid or lymphoid tumour detected in 31 of the 1042 PV patients (3.0%) analyzed from the ECLAP study [51].
Additionally, smoking has been shown to be a significant risk factor for MPN development due in part to the persistent stimulation of myeloid cells [52], with a hazard ratio for daily smokers of 2.5 for any MPN, and of 4.3, 1.8 and 1.7 for MF, ET and PV, respectively [53].
Whether immune dysregulation can be considered an MPN disease “driver” or not remains to be fully elucidated [54], but the connection between chronic inflammation and vascular function, and atherosclerosis in particular, is indisputable [55][56]. For a comprehensive recent review on the role of inflammation in MPN, we refer readers to the article by Di Battista and colleagues published in this special edition [57].
1.4. Molecular Differentiation of PV from Other Haematological Neoplasms
Presence of the JAK2V617F variant, although a distinguishing feature of PV, is not a specific molecular marker for PV. Nor is it a marker for the BCR-ABL1-negative MPN, having been detected in other haematological neoplasms including AML, myelodysplastic syndrome, and chronic myelomonocytic leukaemia [58][59]. The variant has even been found at low levels in the peripheral blood of healthy individuals [60].
One of the first molecular markers identified for the diagnosis of patients with PV was the polycythemia rubra vera 1 gene (PRV1). Granulocytes isolated from the peripheral blood of patients with PV and some cases of ET had an increased expression of PRV1 together with a decreased expression of MPL, with a positive correlation observed between PRV1 expression and splenomegaly, helping to differentiate MPN from secondary erythrocytosis and thrombocytosis [61][62]. However, the use of PRV1 lost popularity when the JAK2V617F driver mutation was discovered in 2005 [7] and PRV1 was not included as a diagnostic marker for PV in the 2008 WHO classification [1]. Moreover, PRV1 expression does not discriminate PV from other BCR-ABL1-negative MPN [63].
A second molecular marker whose expression in circulating mononuclear cells was shown to be significantly increased in 87% of patients with PV but not secondary polycythaemia is the insulin-like growth factor 1 receptor (IGF-1R) [64][65]. The levels of IGF-1R in PV were significantly different in patients with PV compared with patients with ET or PMF, although expression of IGF-1R was significantly higher in patients with the other MPN compared to controls [65].
Distinguishing PV from ET can be complicated due to the shared phenotypic characteristics of the two diseases, particularly the differential diagnosis of PV from JAK2V617F-positive ET. Prior to the 2016 WHO update to the diagnostic criteria for MPN (in which haemoglobin and haematocrit diagnostic thresholds for PV were lowered to 16.5 g/dL and 49% for men, and 16 g/dL and 48% for women, respectively, and bone marrow biopsy was made mandatory, except for cases fulfilling the 2008 WHO diagnostic criteria [2]), PV patients with thrombocytosis at disease presentation were frequently misclassified as JAK2V617F-positive ET patients [66][67][68][69]. It was shown that “masked PV” [68][69] could be distinguished from ET based on red blood cell mass (RCM), with masked PV presenting an increased RCM above 125% [67][70].
This differentiation is important for the prognosis of a patient with MPN, since a higher rate of thrombosis exists among patients with PV than ET, as well as a higher rate of transformation to secondary myelofibrosis (SMF) and AML in PV than ET [68][71]. Importantly, the combined rate of transformation to SMF and/or AML for masked PV was 1.60 per 100 patient years compared to 0.97 for overt PV [71]. These inferior outcomes may possibly be due to a more aggressive form of the disease, the lower intensity of treatment for masked PV patients misdiagnosed with ET, or perhaps due to a later diagnosis [72]. For example, masked PV patients misclassified as JAK2V617F-positive ET would not be phlebotomized. Moreover, misdiagnosed younger patients without prior thrombosis would be categorized as low risk (according to current international guidelines for ET patients) and thus would not even receive cytoreductive treatment, even in those presenting with leucocytosis and thrombocytosis [72].
The differentiation between PV and ET at a molecular level can also be complicated by the presence of passenger mutations in genes common to both MPN. However, mutations in the genes KMT2A (MLL) and TP53 were more common in patients with PV (13.64% and 6.25%, respectively) than patients with PMF (3.45% and 0%, respectively) or ET (0% and 0%, respectively) [73]. Some studies have characterized the expression of several genes and proteins in an attempt to differentiate PV. For example, the overexpression of NFE2 was described in patients with PV and not in patients with ET [74], while high expression of the proteins HSP70 and calreticulin in the membrane of erythrocytes of patients with PV compared with patients with ET suggested an important role for these factors in the proliferation and erythroid function of PV, respectively [75][76]. Moreover, the expression of microRNAs (miRNA), single-stranded non-coding RNA 18–24 nucleotides in length that regulate gene expression by binding to target mRNA, was shown to be different between patients with PV and those with ET (Table 1), as well as between patients with PV and control patients [77][78]. For example, miR-145 and miR-451 were upregulated in nucleated erythroid cells cultured from progenitor cells from patients with PV compared to patients with ET, whereas miR-575 was upregulated and miR-196b was downregulated in PV patients compared to healthy controls [78].
Table 1. Summary of genetic differences between PV and ET that can help differentiate the two MPNs.
| Molecular Marker | PV | ET |
|---|---|---|
| Homozygosity (or high allelic frequency) of JAK2V617F | More common | Less common |
| Mutations in KMT2A and/or TP53 | More common | Less common |
| NFE2 expression | Higher | Lower |
| HSP70 and calreticulin protein expression | Higher | Lower |
| miR-145 and/or miR-451 expression | Higher | Normal |
| miR-222 expression | Lower | Higher |
Nevertheless, the JAK2 allelic burden is sometimes employed to differentiate patients with PV from patients with ET [79][80][81]. Homozygosity of JAK2V617F (represented by a VAF of 50% or more) or a high JAK2V617F allele burden resulting from mitotic recombination of chromosome 9 or 9p loss of heterozygosity [7][36] occurs more frequently in patients with PV while it is significantly less common in patients with ET [80][82]. This observation was confirmed in transgenic mice, in which the increased expression of JAK2V617F resulted in a change of phenotype from ET to PV [83][84].
1.5. Germline Predisposition for Developing PV
A hereditary basis of PV (and other MPNs) was first suggested by observations of a particularly high incidence of PV among people of an Ashkenazi Jewish descent [85], although the common genetic predisposing factor among this population has still yet to be identified. However, the familial basis of PV may be more frequent than previously anticipated, with a population study finding a 5.7-fold increased risk of PV among the first-degree relatives of patients with MPN [86]. Moreover, an interview-based investigation of family history identified familial cases of MPN in 18 of 206 patients with PV (8.7%), with 13 families having first-degree relatives affected by PV [87].
Genetic studies observed that carriers of the JAK2 46/1 haplotype (GGCC) had a higher susceptibility to develop MPN [88][89]. Specifically, carriers of the 46/1 haplotype had a 3.7-fold increased risk of acquiring the JAK2V617F mutation [90], a higher susceptibility to acquire a JAK2 exon 12 mutation [16][91], and an increased disposition (odds ratio of 2.3) for the development of PV versus ET [82].
However, despite the predominance of the JAK2V617F mutation among patients with PV, earlier studies determined that this “driver” mutation was not always disease initiating [92] and might occur as a later secondary genetic event in some patients with MPN [93][94]. Familial cases of MPN, where different family members have a different driver mutation and/or a different MPN, support a pre-JAK2 event and are suggestive of an unknown germline event that predisposes to MPN (unpublished observations, [95]).
One hypothesis is that JAK2 germline mutations, such as JAK2T108A and JAK2L393V, are the genetic predisposing factor that precedes the acquisition of JAK2V617F in MPN [96]. Albeit of uncertain significance, these germline mutations were identified in PV patients as co-existing predisposing mutations with somatic JAK2V617F, and have also been reported in an adenocarcinoma cell line and in diffuse large B cell tumours, respectively, supporting the observation that they provide a proliferative advantage. Nevertheless, their effect on disease initiation has not been fully determined.
Other predisposing germline mutations have been identified among patients with familial MPN, including mutations in the RBBP6 gene, encoding an E3 ubiquitin ligase known to promote the degradation of p53. Variants such as RBBP6R1569H were reported to lie in the p53-binding domain, and thus are predicted to affect p53 function [97]. Other germline predisposition variants for MPN include the single nucleotide polymorphism (SNP) rs2736100 in the gene encoding the telomerase reverse transcriptase TERT [98]. In fact, both the TERT SNP rs2736100 and the JAK2 GGCC haplotype had an additive effect on MPN risk in the presence of an RBBP6 mutation and a higher VAF [99].
Genome-wide association studies (GWAS) have confirmed that various loci associated with telomere length, including rs7705526 and rs2853677, are predisposing factors for MPN risk [100]. Specifically, an increased risk of MPN development was observed for individuals with longer telomere length measured in leukocyte (LTL), as determined using a combined “teloscore” from 11 telomere-length-associated SNPs [101]. The authors of the study concluded that longer telomeres are a risk marker for MPN development.
Nevertheless, a ground-breaking study, presented at this year’s American Society of Hematology (ASH) Annual meeting, demonstrated that driver mutations, such as JAK2V617F were frequently acquired in childhood or even in utero, many decades before an MPN diagnosis [102]. The authors observed a mean time of 34 years between driver mutation acquisition and MPN presentation in patients where JAK2V617F was the only or the first driver mutation. In 4 of the 10 patients analyzed, DNMT3A single-nucleotide variants (SNVs) were the driver event, followed by the acquisition of JAK2V617F, although the DNMT3A mutations were also acquired in childhood or in utero. The authors concluded that the observed latency between driver mutation acquisition and MPN presentation suggests that other factors, including germline, must impact the selection of clones harbouring driver mutations and favor their clonal expansion. Indeed, while normal hematopoietic stem cells accumulated approximately 18 somatic mutations per year, clones harbouring driver mutations such as JAK2V617F accumulated 1.5–5.5-fold more mutations per year and were also found to have shorter telomeres.
1.6. Passenger Mutations
Genetic analyses have led to the description of diverse non-driver mutations in patients with PV, particularly as a result of NGS studies. These include SNVs in genes that encode proteins that regulate epigenetic processes, such as TET2, DNMT3A, ASXL1, EZH2, and IDH1/2, as well as proteins implicated in the splicing of messenger RNA, such as SRSF2, SF3B1 and U2AF1 [9][103], with non-driver mutations detected by NGS in 53% and 75% of PV patients according to two recent studies [104][105].
The effect of passenger mutations on the prognosis of the disease is still not well characterized, although some studies have associated SNVs in ASXL1, SRSF2, and IDH1/2 with poor survival [104][106][107]. Mutations in two novel genes were reported at this year’s ASH Annual meeting to have prognostic value in MPN. SNVs in the gene NFE2 (nuclear factor erythroid 2), with a role in the maturation and differentiation of erythroid cells, were found to be independently associated with shortened time to blastic transformation (with a hazard ratio of 9.9) and overall survival. Such mutations were most common in PV patients compared to PMF and ET, detected in 7.3%, 5.3% and 3.6%, respectively [108]. Similarly, SNVs in the gene STK11 (serine/threonine kinase 11), encoding a kinase with tumour suppressor function, were found to promote leukemic progression in patients with MPN [109].
However, the clinical relevance of some passenger mutations in patients with MPN is still a matter of debate. For instance, the presence of somatic mutations in hematopoietic tissue is positively correlated with age and confers a selective advantage leading to clonal haematopoiesis of indeterminate potential (CHIP, with an established cut-off of ≥2% VAF of in peripheral blood) [82][110][111][112][113][114][115][116].
The most frequently mutated CHIP-associated genes are TET2, DNMT3A, ASXL1 (commonly referred to as the DTA genes), and JAK2. Recent studies have found an association between the accumulation of DTA SNVs and a higher risk for developing myeloid neoplasms, including BCR-ABL1-negative MPN and AML, as well as cardiovascular pathologies and a higher prevalence of developing derived complications from diabetes [116].
TET2 SNVs are particularly common in PV, detected in between approximately 10% and 36% of patients [9][103][105][117][118], and are frequent in both JAK2V617F-positive and -negative MPN. Experiments in mice suggested that mutated TET2 confers a clonal advantage to cells mutated in JAK2 that accelerates their proliferation [119][120]. Moreover, the acquisition of a mutation in DNMT3A followed by mutation in JAK2 was shown to lead to an ET phenotype, whereas the acquisition of the JAK2 mutation first was shown to result in a PV phenotype [121].
Nevertheless, there remains some controversy over whether the presence of CHIP mutations is sufficient for the clonal expansion that results in the emergence of MPN since DTA mutations (as well as the JAK2V617F mutation) are often identified in healthy individuals [113]. An alternative hypothesis is that such mutations simply predispose individuals to acquire mutations in other oncogenes or other genomic abnormalities necessary to drive clonality [102].
Sequencing studies and colony genotyping are helping to determine clonal evolution [122][123] and the order of mutation acquisition. For example, mutations including NFE2, TP53, NRAS, and PPM1D were found to be acquired significantly later in MPN disease [82]. A seminal study by Ortmann et al. investigated the influence of the order of mutations in JAK2 and TET2 in patients with MPN [124]. Firstly, the authors observed that among MPN patients with mutations in both genes, the prior acquisition of TET2 mutations diminished the proliferative effect of the JAK2V617F mutation. Secondly, they found that PV was more common among JAK2-first patients compared to ET or PMF. Finally, they concluded that the order of mutation acquisition influenced the age at which a patient presented with an MPN, with JAK2-first patients presenting at a younger age compared with TET2-first MPN patients. This observation was supported by a recent study which concluded that the order of mutation acquisition was associated with time to MPN presentation [102].
2. Prognosis of PV
Patients with PV have a median overall survival of 13.5 years [125]. At diagnosis, PV patients are classified into groups of high, intermediate or low risk for overall survival according to the clinical variables of patient age, leukocyte count and history of venous thromboembolic events (Table 2) [126].
Table 2. Stratification of risk of survival in polycythaemia vera according to the classical risk score.
| Risk Factor | Score |
|---|---|
| Age ≥ 67 years | 5 |
| Age 57–66 years | 2 |
| Leukocytes ≥ 15 × 109/L | 1 |
| Venous thrombosis | 1 |
Low risk: score 0, intermediate risk: score 1 or 2, high risk: score ≥ 3 (adapted from [126]).
Until very recently there were no well-characterized molecular determinants of prognosis that could be used in the clinic to predict the survival of patients with PV [4], with the ELN score only considering patient age and history of thrombosis [18]. However, the recently proposed Mutation-enhanced International Prognostic Scoring System (MIPSS-PV) includes the mutation SRSF2 due to its association with worse overall survival (Table 3) [127] to classify patients into low-risk (score of 0–1), intermediate-risk (score of 2–3) or high-risk (score ≥ 4) groups.
Table 3. Stratification of risk of survival in polycythaemia vera according to the Mutation-enhanced International Prognostic Scoring System (MIPSS-PV) (adapted from [127]).
| Risk Factor | Score |
|---|---|
| Age ≥ 67 years | 2 |
| Leukocytes ≥ 15 × 109/L | 1 |
| Thrombosis history | 1 |
| SRSF2 mutation | 3 |
2.1. JAK2V617F Allelic Burden
Although quantification of the JAK2V617F allelic burden is not obligatory for the diagnosis and follow-up of patients with PV according to the WHO criteria [1][2], patients homozygous for JAK2V617F were shown to be more symptomatic than patients who were heterozygous. Specifically, a positive correlation was described between a high allelic frequency of mutated JAK2 and clinical presentation including pruritus [128][129][130], myelopoiesis [8][129], splenomegaly [8][128][129][131][132], and a negative correlation with the platelet count [8][129][132][133].
Moreover, determination of the JAK2V617F VAF at diagnosis can provide important prognostic information. For example, in PV, a higher mutant allele burden has been associated with an increased incidence of progression to SMF [8][80][128][134].
Both of these observations are suggestive of a “dosage” effect (reviewed in [95]), where a high JAK2V617F burden results in a stronger activation of JAK2/STAT (and possibly other) signalling pathways, leading to the stronger activation of downstream genes [135], and thus a more aggressive phenotype. In support of this hypothesis, the expression level of several JAK2 target genes—such as PRV1 [135] and NFE2 [136]—was shown to be dose dependent for the mutant allele burden.
A high JAK2V617F allele burden in patients with PV has also been associated with a risk of thrombotic events [137][138][139]. A study of 1306 patients with MPN including 397 patients with PV, observed an incidence of thrombosis at a diagnosis of 21% for homozygous and 15% for heterozygous JAK2V617F [139], with JAK2V617F homozygosity at diagnosis identified as an independent risk factor for major vascular events in a multivariate analysis [138]. A second study reported a 7.1-fold increased risk of major vascular events for patients with a mutant allele of 75% and above compared to less than 25% VAF during follow-up [127]. However, one study associated a significantly increased risk of venous thromboembolism with a much lower mutant allele burden, of just 20% or more [128]. Despite the differences in the VAF threshold reported to have a negative effect on thrombotic risk, it is clear that patients with a higher JAK2 mutant burden are at risk of both vascular events and fibrotic transformation.
Nevertheless, one study’s findings contradict the prognostic impact of the JAK2V617F allelic burden in patients with PV. Gene expression profiling of CD34+ cells isolated from 19 patients with PV led to the identification of a group of patients with a more aggressive disease—with higher rates of blastic transformation and worse overall survival—and a group with a less aggressive form despite similar JAK2V617F allele frequencies in both groups. Patients in the group with more aggressive PV showed differential expression of the NOTCH and SHH pathways as well as inflammatory cytokines and histone genes [140]. Interestingly, this study also revealed gender-specific differences in the expression profile in that men with PV had significantly more differentially regulated genes than women with PV.
2.2. Cytogenetics
Cytogenetic abnormalities are detected in approximately 10–20% of PV patients at diagnosis, with some of the most common alterations including gain of chromosomes 8 and 9, and deletion of (1p), (13q) and (20q) [141][142][143].
Although some studies have not shown a prognostic difference based on cytogenetic characteristics [143], several groups have reported, as association between a higher risk of fibrotic progression and the presence of chromosome 12 abnormalities [144], the gain of (1q) [145], and an abnormal karyotype. Moreover, the majority of investigations, including one by the International Working Group for Neoplasms Research and Treatment (IWG-MRT), have reported an association between an abnormal karyotype and transformation to myelodysplastic syndrome or AML [143][145][146][147] and poorer overall survival [142][145][147].
Interestingly, a retrospective study of 422 PV patients with cytogenetic information available at diagnosis observed dynamic changes associated with progression to AML, including an increased frequency of abnormal karyotype, increasing from 20% to 90% in the blast phase, as well as a different distribution of cytogenetic abnormalities [146]. For example, complex karyotype as well as deletion of (5q)/chromosome 5, 7q/chromosome 7, and (17p)/chromosome 17/i(17q) were more common in the blast phase, whereas the gain of chromosomes 8 and 9 were more common in the chronic phase of the disease. Using this information, the authors stratified patients into low (normal karyotype; +8, +9; or presence of one other alteration), intermediate (del(20q), +(1q) or presence of two other alterations) and high-risk (complex karyotype) groups according to their cytogenetics at diagnosis [145].
2.3. Thrombosis
The BCR-ABL1-negative MPNs PV, ET and PMF are associated with a high frequency of haemorrhages and thrombosis, including myocardial infarction, ischemic stroke, deep vein thrombosis, and thrombo- and pulmonary embolisms. Notably, vascular events are the major cause of morbimortality among patients with PV, with the incidence of events estimated between 6% and 17% over three years [148][149].
Thrombotic risk prediction tools have been developed for each BCR-ABL1-negative MPN subtype. However, thrombotic events are frequent among patients with PV, even among those classified as low-risk according to the current ELN recommendations [18][150], as well as patients who receive cytoreductive or anti-aggregant treatment [151].
Several common thrombophilia markers are associated with an increased risk of developing vascular events in patients with PV (reviewed in [152]), including high levels of C-reactive protein in blood serum and SNVs in the factor V-Leiden (F5) gene [153][154][155]. Higher factor VIII levels, a known risk factor for venous thrombosis and coronary artery disease [156], were identified in PV patients compared to healthy subjects, with levels of 141 versus 98 IU/dL, respectively [157]. The PV patients were also found to have significantly higher VWF antigen and activity which, by multivariate analysis, was predicted for by JAK2V617F allelic burden. Nevertheless, the ELN does not currently recommend thrombophilia testing as part of routine clinical practice in PV or MPN patients [18][150].
Other prognostic biomarkers reported for PV include an abnormal karyotype, identified as a risk factor for venous thrombosis [142], and leucocytosis, a risk factor for arterial thrombosis, although its impact on venous thrombosis remains inconclusive [158][159].
The International Prognostic Score of thrombosis in World Health Organization-essential thrombocythemia (IPSET-thrombosis) was the first prognostic algorithm to incorporate the JAK2V617F mutation to better predict the risk of thrombosis among ET patients [160], although the influence of JAK2V617F in PV patients as well as the clinical significance of the allelic burden on thrombotic risk remain to be determined. Some studies have found that a higher JAK2V617F allelic burden was associated with increased risk of thrombosis in PV and ET [161]. For example, JAK2V617F allele burden cut-offs for PV > 25.7% and > 90.4% were established for arterial thrombosis and for venous thrombosis, respectively [162].
Moreover, elevated levels of the inflammatory biomarker C-reactive protein in peripheral blood were associated with the PV phenotype (vs. essential thrombocythemia), older age, cardiovascular risk factors and a JAK2V617F allele burden over 50% [56][163], further supporting suggestions of a possible link between inflammation and atherosclerosis [55][56][57]. A seminal study from 2017 identified an association between mutations in the DTA and JAK2 genes and coronary disease [116]. Mechanistically, mutations to DTA genes are suggested to alter methylation patterns and thus cause an increased transcription of pro-inflammatory genes triggering atherosclerosis [116][164][165]. To support this theory, Fuster and colleagues studied the effect of the expansion of TET2-deficient cells in atherosclerosis-prone, low-density lipoprotein receptor-deficient mice and observed that the partial reconstitution of the bone marrow with TET2-deficient cells was sufficient to generate clonal expansion, which was also associated with a marked increase in the size of atheromatous plaques and the increased secretion of pro-inflammatory cytokines such as IL-1β [164].
In accordance with these results, Jaiswal et al. found that individuals with clonal hematopoiesis had a 1.9-fold higher risk of developing coronary disease and a 4-fold higher risk of myocardial infarction, rising to 12.1-fold in individuals with the JAK2V617F mutation [116]. In support of the link between constitutive JAK2 activation and increased risk of vascular events, the rs3184504 SNP in the SH2B3 (LNK) gene was also associated with an increased risk of coronary heart disease [166]. This variant, encoding the TT genotype (R262W), caused an increased platelet count, leukocytosis and hypertension. At the molecular level, SH2B3 mutation increased megakaryopoiesis via the upregulation of AKT signalling, shown to be specifically upregulated in platelets and to promote prothrombotic and proatherogenic aggregates [167]. Specifically, our group recently showed that DTA mutations were associated with increased thrombotic risk in patients with PV, with a specific interaction identified for pathogenic TET2 mutations [105].
Activated leukocytes (including neutrophils and monocytes) appear to play an important role in the development of platelet aggregates, markers of a prothrombotic state. In addition, there are reports that leuko-platelet aggregations directly correlate with platelet and leukocyte counts [168][169], adding weight to leukocytosis as a thrombotic risk factor. Interestingly, and as previously mentioned, the expression of CD177 (PRV-1), a GPI-linked cell surface glycoprotein with a role in neutrophil activation, was increased in PV patients, and to a lesser extent in ET and PMF patients [61][62]. Indeed, higher CD177 expression was associated with an increased risk of thrombotic and bleeding complications due to increased circulating neutrophils [170][171]. For a complete and recent review on the role of neutrophils in thrombosis, we refer readers to the article by Ferrer-Marín and colleagues published in this special edition [172]. It is clear that we need to further elucidate the molecular mechanisms of leukocyte–platelet interactions in order to design better prophylactic treatment plans for the prevention of thrombotic complications in patients with PV.
2.4. Molecular Indicators of Transformation
Progression to fibrotic disease (post-PV MF) or blastic disease, both associated with dismal survival rates, is estimated to occur in 10% and 15% of PV patients, respectively [173]. Molecular indicators of fibrotic progression from PV are not well characterized. Some evidence exists suggesting an association between chromosome 12 abnormalities and an increased risk of developing post-PV MF [174]. Evidence also exists to support a significantly higher JAK2V617F allelic frequency [8][80][128][134][174] or a progressive increase [175] in patients who transform to SMF. Unlike in PMF, the presence of additional high-risk mutations (ASXL1, EZH2, IDH1, IDH2 or SRSF2, as single or multiple mutations) was not correlated with reduced leukaemia-free survival or indeed overall survival, with the exception of mutations in SRSF2, associated with reduced survival [176][177].
As the molecular profile of post-PV MF was shown to be considerably different from PMF [178], the Myelofibrosis Secondary to PV and ET Prognostic Model (MYSEC-PM) was specifically developed to predict survival in post-PV/post-ET MF (Table 4) [179].
Table 4. Stratification of risk of survival in post-PV MF according to the Myelofibrosis Secondary to PV and ET Prognostic Model (MYSEC-PM) (adapted from [179]). Patients are classified into low (score 0–10), intermediate 1 (score 11–13), intermediate 2 (score 14–15) or high (score > 16) risk groups. The MYSEC-PM is available online as a risk calculator (http://www.mysec-pm.eu/, accessed on 2 November 2020).
| Risk Factor | Score |
|---|---|
| Haemoglobin < 11 g/dL | 2 |
| Circulating blasts ≥3% | 2 |
| CALR-unmutated 1 | 2 |
| Platelets < 150 × 109/L | 1 |
| Constitutional symptoms | 1 |
| Age | 0.15 per year |
1 For patients with post-ET MF.
The score was shown to more effectively assess prognosis in post-PV MF patients, with poor agreement for risk classification between the DIPSS and MYSEC-PM scores when applied to the same post-PV patients [178][180]. Therefore, the MYSEC-PM is the most appropriate prognostic score for post-PV MF patients rather than the International Prognostic Scoring System (IPSS) or Dynamic IPSS (DIPSS) scores, both of which were developed for PMF.
Interestingly, in a retrospective study of 376 patients with SMF, including 188 post-PV MF, monosomal karyotype (observed in 8.5% of the SMF patients studied) was associated with significantly worse survival, with a median of 2 years, independently of the MYSEC-PM risk group [181].
In terms of blastic transformation, it is also clear that the genetic profile of patients with post-PV AML differs from that of de novo AML. For example, mutations in FLT3 and NPM1 are very uncommon and the JAK2V617F-positive clone is frequently lost in transformation to AML [182][183]. Moreover, abnormal karyotype is a risk factor for leukemic transformation in PV and ET [142][143][144][147], estimated to occur in 80% of PV cases that progress [143].
In some cases, the transformation from PV to AML is associated with the loss of function of TP53 due to an acquired mutation in the TP53 gene leading to clonal dominance [82][175][184][185][186]. In fact, in a mouse model, the expression of JAK2V617F in combination with the loss of TP53 was demonstrated to cause an AML phenotype [186]. This result was reflected in a later NGS study, which observed that post-PV/-ET AML patients with a TP53 mutation had a survival rate at 12 months of just 18% compared to 48% for patients without a TP53 mutation [185].
Other mutations associated with a higher risk of leukemic transformation include ASXL1, EZH2, RUNX1 and SRSF2 [82][127][185][187]. Indeed, a recent French study that investigated the time between diagnosis and leukemic progression in 49 patients with post-PV/ET AML (including 24 post-PV) observed that mutations in IDH1/2, RUNX1, and U2AF1 were associated with a shorter time to transformation, while mutations in the genes TP53, NRAS, and BCORL1 were associated with a longer time to transformation [185]. Such mutations are often detectable at low allelic frequencies in earlier stages of the disease, while others may appear later in the progression [184][185][186].
TET2 was also observed in some studies to be a gene whose mutation is associated with the leukemic transformation of PV patients. One study that investigated mutations in 63 patients with AML secondary to an MPN found that mutations in TET2 were acquired at transformation in 43% of cases [188], and the acquisition of mutations in TET2 in patients with PV JAK2V617F was associated with transformation to AML and reduced survival [117]. Nevertheless, the prognostic impact of TET2 mutations, and indeed other mutations associated with CHIP, is not entirely clear. For example, when the impact of additional mutations on the risk of leukemic transformation was investigated, the presence of >1 additional SNV was associated with a higher risk of leukemic transformation and this association was even higher when TET2 SNVs were removed [185]. Moreover, mutations in ASXL1 were observed at similar allelic frequencies before and after transformation, suggesting that they may not contribute to the dominance of the emerging leukemic clone [186].
One of the largest sequencing studies performed to date, on 2035 MPN patients (including 356 PV patients), identified eight genetic subgroups [82]. The authors developed a sophisticated prognostic model based on 63 clinical and genomic variables to estimate a patient’s probability of leukemic transformation. These findings have now been adapted into a personalized risk calculator, available online (https://cancer.sanger.ac.uk/mpn-multistage/, accessed on 26 November 2020).
3. Treatment
Thrombosis is the most common cause of morbidity and death, followed by the complications of myelofibrosis and development of leukaemia. As such, the primary treatment goals in MPN are to avoid thrombosis and bleeding and minimize transformation to myelofibrosis or AML while treating MPN-related symptoms to improve the quality of life of patients with PV [3][4][189].
Frontline strategies to prevent vascular events include prophylactic aspirin with phlebotomies as and when required to maintain haematocrit (HCT) levels below 45%. In addition, patients often require cytoreductive treatment, most commonly hydroxyurea (HU), according to recent European guidelines [3][4][18][150]. However, approximately 15–20% of patients develop intolerance or resistance to HU with continued use [190][191].
To date, there is very little information on how to identify patients with PV at risk of developing resistance to HU treatment. However, one recent study used NGS to molecularly characterize 37 PV patients with resistance or intolerance to HU [192]. The authors found that patients with a homozygous JAK2 mutation had a higher risk of developing resistance after 5 years of HU treatment compared to those with a heterozygous JAK2 mutation (27% vs. 14.5%), while for those with a TP53 alteration (mutation or copy number alteration) the probability increased to 64% [192].
For HU-resistant or -intolerant patients, ruxolitinib is the only JAK2 inhibitor currently approved by the European Medicines Agency (EMA) and the Food and Drug Administration (FDA) as second-line treatment [193][194], following the positive results of the clinical trials RESPONSE, in PV patients with splenomegaly (NCT01243944, ClinicalTrials.gov) [195], and RESPONSE-2, in PV patients without splenomegaly [196]. Disappointingly, a phase 2 study with momelotinib, another JAK2 inhibitor, in patients with PV (NCT019898828, ClinicalTrials.gov) was terminated early due to low efficacy [197].
One particularly positive observation from clinical trials with ruxolitinib was the reduction in the incidence of thromboembolic events, 5-fold lower in the group of patients receiving ruxolitinib in comparison with the group receiving the best available treatment (BAT; HU in the majority of patients) in Week 32 of treatment, and 4.5-fold lower in Week 80, although this was not a primary study objective [193][194]. These observations were confirmed by a meta-analysis which included data from 750 PV and MF patients, including those who participated in the RESPONSE trial [198]. In addition, the highly anticipated results of the MAJIC clinical trial (EudraCT 201100527918) should provide more information on the frequency of thrombotic events in patients with PV treated with ruxolitinib.
Also noteworthy is the recent European Medicine Agency (EMA) approval of Ropeginterferon alpha-2b (Besremi®) for the treatment of adult patients with polycythaemia vera [199] as a result of the PROUD, CONTINUATION-PV [200] and Low-PV clinical trials [201]. Besremi® can be used as first-line treatment but may also offer an alternative for HU- and/or ruxolitinib-resistant or -intolerant patients.
Ropeginterferon alfa-2b is a monopegylated interferon (IFN) that can be administered once every 2 weeks. The molecule binds to interferon α/β receptors (IFNAR) of endogenous interferon cytokines on the cellular membrane of many blood cells, including B cells, NK cells, mononuclear cells and macrophages, as well as endothelial cells, epithelial cells, hepatocytes and many other cell types [202]. Although the mechanism of action of IFN remains to be elucidated, it does have pro-apoptotic properties, activating the expression of caspase and tumour necrosis factor-related apoptosis-inducing ligand (TRAIL) [203].
Advantages of IFN include that it is not leukomogenic in comparison to HU [199][204], and its long-term use (>18 months) leads to a sustained decrease in JAK2 allelic burden in the majority of patients (Figure 2) [199]. Nevertheless, the clinical relevance of this reduction remains to be determined [199]. For example, is the reduction in JAK2 allelic burden reflected in the patient’s response to therapy with a reduction in symptom burden? A lower incidence of fibrotic or leukemic transformation? Improved overall survival?
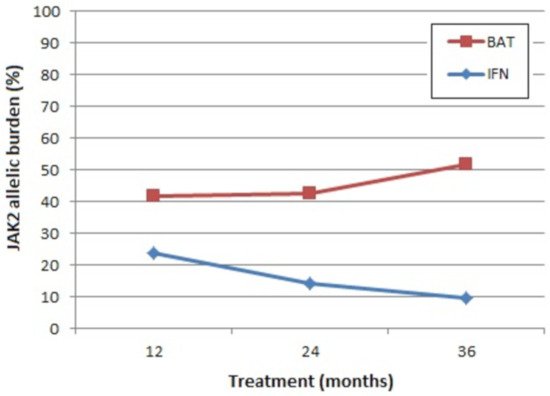
Figure 2. Mean absolute levels of JAK2 allelic burden. BAT: best available therapy (100% of the patients received hydroxyurea at least once), IFN: pegylated interferon-alpha 2b. Representation of results from CONTINUATION-PV (adapted from [199]).
Other areas of doubt in relation to treatment with IFN include its effect on thrombosis. One recent study observed the significant elevation of the pro-coagulant biomarkers thrombin, fibrinogen, Von Willebrand factor, shear-induced platelet aggregation, and factor VIII coagulant activity (FVIII:C) in the blood plasma of patients with PV or ET treated with IFN compared to those treated with HU or no treatment, independently of gender and patient age [205]. Whether these reported effects of IFN treatment on the haemostatic profile of PV patients are associated with an increased thrombotic risk remains to be determined; however, important differences were reported between the risk of thromboembolic events for PV patients treated with IFN (8.7%, 11/127) versus best-available therapy (BAT, 5.5%, 7/127) [199].
The approval of Besremi® is the first new treatment option for patients with PV in many years, although several others are in different stages of development and have shown promising preliminary results, such as the histone deacetylase inhibitor givinostat and the hepcidin-mimetic PTG-300. Treatment strategies are outside the scope of this review on the molecular-informed diagnosis and prognosis of patients with PV; thus, we refer readers to a comprehensive review of current treatment options as well as novel agents in a recent volume of International Journal of Molecular Sciences [206].
References
- Swerdlow, S.H.; Campo, E.; Harris, N.L.; Jaffe, E.S.; Pileri, S.A.; Stein, H.; Thiele, J. World Health Organization Classification of Tumours of Haematopoietic and Lymphoid Tissues. In World Health Organization Classification of Tumours, 4th ed.; Bosman, F.T., Jaffe, E.S., Lakhani, S.R., Ohgaki, H., Eds.; IARC: Lyon, France, 2008; Volume 2.
- Arber, D.A.; Orazi, A.; Hasserjian, R.; Thiele, J.; Borowitz, M.J.; Le Beau, M.M.; Bloomfield, C.D.; Cazzola, M.; Vardiman, J.W. The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood 2016, 127, 2391–2405.
- Raedler, L.A. Diagnosis and Management of Polycythemia Vera: Proceedings from a Multidisciplinary Roundtable. Am. Health Drug Benefits 2014, 7, S36–S47.
- Vannucchi, A.M.; Barbui, T.; Cervantes, F.; Harrison, C.; Kiladjian, J.J.; Kröger, N.; Thiele, J.; Buske, C.; ESMO Guidelines Committee. Philadelphia chromosome-negative chronic myeloproliferative neoplasms: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann. Oncol. 2015, 26, 85–99.
- Nangalia, J.; Grinfeld, J.; Green, A.R. Pathogenesis of Myeloproliferative Disorders. Annu Rev. Pathol Mech Dis. 2016, 11, 101–126.
- Baxter, E.J.; Scott, L.M.; Campbell, P.J.; East, C.; Fourouclas, N.; Swanton, S.; Vassiliou, G.S.; Bench, A.J.; Boyd, E.M.; Curtin, N.; et al. Acquired mutation of the tyrosine kinase JAK2 in human myeloproliferative disorders. Lancet 2005, 365, 1054–1061.
- Kralovics, R.; Passamonti, F.; Buser, A.S.; Teo, S.S.; Tiedt, R.; Passweg, J.R.; Tichelli, A.; Cazzola, M.; Skoda, R.C. A gain-of-function mutation of JAK2 in myeloproliferative disorders. N. Engl. J. Med. 2005, 352, 1779–1790.
- Passamonti, F.; Rumi, E.; Pietra, D.; Elena, C.; Boveri, E.; Arcaini, L.; Roncoroni, E.; Astori, C.; Merli, M.; Boggi, S.; et al. A prospective study of 338 patients with polycythemia vera: The impact of JAK2 (V617F) allele burden and leukocytosis on fibrotic or leukemic disease transformation and vascular complications. Leukemia 2010, 24, 1574–1579.
- Langabeer, S.E.; Andrikovics, H.; Asp, J.; Bellosillo, B.; Carillo, S.; Haslam, K.; Kjaer, L.; Lippert, E.; Mansier, O.; Oppliger Leibundgut, E.; et al. Molecular diagnostics of myeloproliferative neoplasms. Eur. J. Haematol. 2015, 95, 270–279.
- Broséus, J.; Park, J.H.; Carillo, S.; Hermouet, S.; Girodon, F. Presence of calreticulin mutations in JAK2-negative polycythemia vera. Blood 2014, 124, 3964–3966.
- Belcic Mikic, T.; Pajic, T.; Sever, M. CALR mutations in a cohort of JAK2 V617F negative patients with suspected myeloproliferative neoplasms. Sci. Rep. 2019, 9, 19838.
- Gong, J.Z.; Cook, J.R.; Greiner, T.C.; Hedvat, C.; Hill, C.E.; Lim, M.S.; Longtine, J.A.; Sabath, D.; Wang, Y.L.; Association for Molecular Pathology. Laboratory practice guidelines for detecting and reporting JAK2 and MPL mutations in myeloproliferative neoplasms: A report of the Association for Molecular Pathology. J. Mol. Diagn 2013, 15, 733–744.
- Passamonti, F.; Elena, C.; Schnittger, S.; Skoda, R.C.; Green, A.R.; Girodon, F.; Kiladjian, J.J.; McMullin, M.F.; Ruggeri, M.; Besses, C.; et al. Molecular and clinical features of the myeloproliferative neoplasm associated with JAK2 exon 12 mutations. Blood 2011, 117, 2813–2816.
- Li, S.; Kralovics, R.; De Libero, G.; Theocharides, A.; Gisslinger, H.; Skoda, R.C. Clonal heterogeneity in polycythemia vera patients with JAK2 exon12 and JAK2-V617F mutations. Blood 2008, 111, 3863–3866.
- Scott, L.M.; Tong, W.; Levine, R.L.; Scott, M.A.; Beer, P.A.; Stratton, M.R.; Futreal, P.A.; Erber, W.N.; McMullin, M.F.; Harrison, C.N.; et al. JAK2 exon 12 mutations in polycythemia vera and idiopathic erythrocytosis. N. Engl. J. Med. 2007, 356, 459–468.
- Pietra, D.; Li, S.; Brisci, A.; Passamonti, F.; Rumi, E.; Theocharides, A.; Ferrari, M.; Gisslinger, H.; Kralovics, R.; Cremonesi, L.; et al. Somatic mutations of JAK2 exon 12 in patients with JAK2 (V617F)-negative myeloproliferative disorders. Blood 2008, 111, 1686–1689.
- Grisouard, J.; Li, S.; Kubovcakova, L.; Rao, T.N.; Meyer, S.C.; Lundberg, P.; Hao-Shen, H.; Romanet, V.; Murakami, M.; Radimerski, T.; et al. JAK2 exon 12 mutant mice display isolated erythrocytosis and changes in iron metabolism favoring increased erythropoiesis. Blood 2016, 128, 839–851.
- Barbui, T.; Tefferi, A.; Vannucchi, A.M.; Passamonti, F.; Silver, R.T.; Hoffman, R.; Verstovsek, S.; Mesa, R.; Kiladjian, J.J.; Hehlmann, R.; et al. Philadelphia chromosome-negative classical myeloproliferative neoplasms: Revised management recommendations from European LeukemiaNet. Leukemia 2018, 32, 1057–1069.
- Jovanovic, J.V.; Ivey, A.; Vannucchi, A.M.; Lippert, E.; Oppliger Leibundgut, E.; Cassinat, B.; Pallisgaard, N.; Maroc, N.; Hermouet, S.; Nickless, G.; et al. Establishing optimal quantitative-polymerase chain reaction assays for routine diagnosis and tracking of minimal residual disease in JAK2-V617F-associated myeloproliferative neoplasms: A joint European LeukemiaNet/MPN&MPNr-EuroNet (COST action BM0902) study. Leukemia 2013, 2, 2032–2039.
- Link-Lenczowska, D.; Pallisgaard, N.; Cordua, S.; Zawada, M.; Czekalska, S.; Krochmalczyk, D.; Kanduła, Z.; Sacha, T. A comparison of qPCR and ddPCR used for quantification of the JAK2 V617F allele burden in Ph negative MPNs. Ann. Hematol. 2018, 97, 2299–2308.
- Coccaro, N.; Tota, G.; Anelli, L.; Zagaria, A.; Specchia, G.; Albano, F. Digital PCR: A Reliable Tool for Analyzing and Monitoring Hematologic Malignancies. Int. J. Mol. Sci. 2020, 21, 3141.
- Ma, W.; Kantarjian, H.; Zhang, X.; Yeh, C.H.; Zhang, Z.J.; Verstovsek, S.; Albitar, M. Mutation profile of JAK2 transcripts in patients with chronic myeloproliferative neoplasias. J. Mol. Diagn 2009, 11, 49–53.
- Maslah, N.; Verger, E.; Schlageter, M.H.; Miclea, J.M.; Kiladjian, J.J.; Giraudier, S.; Chomienne, C.; Cassinat, B. Next-generation sequencing for JAK2 mutation testing: Advantages and pitfalls. Ann. Hematol. 2018, 98, 111–118.
- Singh, R.R. Next-Generation Sequencing in High-Sensitive Detection of Mutations in Tumors: Challenges, Advances, and Applications. J. Mol. Diagn 2020, 22, 994–1007.
- Hocking, J.; Mithraprabhu, S.; Kalff, A.; Spencer, A. Liquid biopsies for liquid tumors: Emerging potential of circulating free nucleic acid evaluation for the management of hematologic malignancies. Cancer Biol. Med. 2016, 13, 215–225.
- Garcia-Gisbert, N.; Fernández-Ibarrondo, L.; Fernández-Rodríguez, C.; Gibert, J.; Andrade-Campos, M.; Arenillas, L.; Camacho, L.; Angona, A.; Longarón, R.; Salar, A.; et al. Circulating cell-free DNA improves the molecular characterisation of Ph-negative myeloproliferative neoplasms. Br. J. Haematol. 2020.
- Lasho, T.L.; Pardanani, A.; Tefferi, A. LNK mutations in JAK2 mutation-negative erythrocytosis. N. Engl. J. Med. 2010, 363, 1189–1190.
- Oh, S.T.; Simonds, E.F.; Jones, C.; Hale, M.B.; Goltsev, Y.; Gibbs, K.D., Jr.; Merker, J.D.; Zehnder, J.L.; Nolan, G.P.; Gotlib, J. Novel mutations in the inhibitory adaptor protein LNK drive JAK-STAT signaling in patients with myeloproliferative neoplasms. Blood 2010, 116, 988–992.
- Ema, H.; Sudo, K.; Seita, J.; Matsubara, A.; Morita, Y.; Osawa, M.; Takatsu, K.; Takaki, S.; Nakauchi, H. Quantification of self-renewal capacity in single hematopoietic stem cells from normal and Lnk-deficient mice. Dev. Cell 2005, 8, 907–914.
- Bersenev, A.; Wu, C.; Balcerek, J.; Tong, W. Lnk controls mouse hematopoietic stem cell self-renewal and quiescence through direct interactions with JAK2. J. Clin. Invest 2008, 118, 2832–2844.
- Guglielmelli, P.; Pietra, D.; Pane, F.; Pancrazzi, A.; Cazzola, M.; Vannucchi, A.M.; Tura, S.; Barosi, G. Recommendations for molecular testing in classical Ph1-neg myeloproliferative disorders-A consensus project of the Italian Society of Hematology. Leuk Res. 2017, 58, 63–72.
- Chen, Y.; Fang, F.; Hu, Y.; Liu, Q.; Bu, D.; Tan, M.; Wu, L.; Zhu, P. The Polymorphisms in LNK Gene Correlated to the Clinical Type of Myeloproliferative Neoplasms. PLoS ONE 2016, 11, e0154183.
- Parganas, E.; Wang, D.; Stravopodis, D.; Topham, D.J.; Marine, J.C.; Teglund, S.; Vanin, E.F.; Bodner, S.; Colamonici, O.R.; van Deursen, J.M.; et al. Jak2 is essential for signaling through a variety of cytokine receptors. Cell 1998, 93, 385–395.
- Quelle, F.W.; Wang, J.; Feng, J.; Wang, D.; Cleveland, J.L.; Ihle, J.N.; Zambetti, G.P. Cytokine rescue of p53-dependent apoptosis and cell cycle arrest is mediated by distinct Jak kinase signaling pathways. Genes Dev. 1998, 12, 1099–1107.
- Levine, R.L.; Wadleigh, M.; Cools, J.; Ebert, B.L.; Wernig, G.; Huntly, B.J.; Boggon, T.J.; Wlodarska, I.; Clark, J.J.; Moore, S.; et al. Activating mutation in the tyrosine kinase JAK2 in polycythemia vera, essential thrombocythemia, and myeloid metaplasia with myelofibrosis. Cancer Cell 2005, 7, 387–397.
- Kralovics, R.; Guan, Y.; Prchal, J.T. Acquired uniparental disomy of chromosome 9p is a frequent stem cell defect in polycythemia vera. Exp. Hematol. 2002, 30, 229–236.
- Jamieson, C.H.; Gotlib, J.; Durocher, J.A.; Chao, M.P.; Mariappan, M.R.; Lay, M.; Jones, C.; Zehnder, J.L.; Lilleberg, S.L.; Weissman, I.L. The JAK2 V617F mutation occurs in hematopoietic stem cells in polycythemia vera and predisposes toward erythroid differentiation. Proc. Natl. Acad. Sci. USA 2006, 103, 6224–6229.
- Laubach, J.P.; Fu, P.; Jiang, X.; Salter, K.H.; Potti, A.; Arcasoy, M.O. Polycythemia vera erythroid precursors exhibit increased proliferation and apoptosis resistance associated with abnormal RAS and PI3K pathway activation. Exp. Hematol. 2009, 37, 1411–1422.
- Walz, C.; Crowley, B.J.; Hudon, H.E.; Gramlich, J.L.; Neuberg, D.S.; Podar, K.; Griffin, J.D.; Sattler, M. Activated Jak2 with the V617F point mutation promotes G1/S phase transition. J. Biol. Chem. 2006, 281, 18177–18183.
- Plo, I.; Nakatake, M.; Malivert, L.; de Villartay, J.P.; Giraudier, S.; Villeval, J.L.; Wiesmuller, L.; Vainchenker, W. JAK2 stimulates homologous recombination and genetic instability: Potential implication in the heterogeneity of myeloproliferative disorders. Blood 2008, 112, 1402–1412.
- Marty, C.; Lacout, C.; Droin, N.; Le Couédic, J.P.; Ribrag, V.; Solary, E.; Vainchenker, W.; Villeval, J.L.; Plo, I. A role for reactive oxygen species in JAK2 V617F myeloproliferative neoplasm progression. Leukemia 2013, 27, 2187–2195.
- Chen, E.; Ahn, J.S.; Massie, C.E.; Clynes, D.; Godfrey, A.L.; Li, J.; Park, H.J.; Nangalia, J.; Silber, Y.; Mullally, A.; et al. JAK2V617F promotes replication fork stalling with disease-restricted impairment of the intra-S checkpoint response. Proc. Natl. Acad. Sci. USA 2014, 111, 15190–15195.
- Nakatake, M.; Monte-Mor, B.; Debili, N.; Casadevall, N.; Ribrag, V.; Solary, E.; Vainchenker, W.; Plo, I. JAK2(V617F) negatively regulates p53 stabilization by enhancing MDM2 via La expression in myeloproliferative neoplasms. Oncogene 2012, 31, 1323–1333.
- Silva, M.; Richard, C.; Benito, A.; Sanz, C.; Olalla, I.; Fernández-Luna, J.L. Expression of Bcl-x in erythroid precursors from patients with polycythemia vera. N. Engl. J. Med. 1998, 338, 564–571.
- Zhao, R.; Follows, G.A.; Beer, P.A.; Scott, L.M.; Huntly, B.J.; Green, A.R.; Alexander, D.R. Inhibition of the Bcl-xL deamidation pathway in myeloproliferative disorders. N. Engl. J. Med. 2008, 359, 2778–2789.
- Scherber, R.; Geyer, H.; Dueck, A.; Johnston, C.; Langlais, B.; Padrnos, L.; Palmer, J.; Fleischman, A.; Mesa, R. Nutritional needs and preferences of myeloproliferative neoplasm patients: Phase IA of the Nutrient Study; European Hematology Association Congress: Madrid, Spain, 2017.
- Coussens, L.M.; Web, Z. Inflammation and cancer. Nature 2002, 420, 860–867.
- Colotta, F.; Allavena, P.; Sica, A.; Garlanda, C.; Mantovani, A. Cancer-related inflammation, the seventh hallmark of cancer: Links to genetic instability. Carcinogenesis 2009, 30, 1073–1081.
- Marchetti, M.; Ghirardi, A.; Masciulli, A.; Carobbio, A.; Palandri, F.; Vianelli, N.; Rossi, E.; Betti, S.; Di Veroli, A.; Iurlo, A.; et al. Second cancers in MPN: Survival analysis from an international study. Am. J. Hematol. 2020, 95, 295–301.
- Pettersson, H.; Knutsen, H.; Holmberg, E.; Andréasson, B. Increased incidence of another cancer in myeloproliferative neoplasms patients at the time of diagnosis. Eur. J. Haematol. 2015, 94, 152–156.
- Ghirardi, A.; Carobbio, A.; Masciulli, A.; Barbui, T. Incidence of solid tumors in polycythemia vera treated with phlebotomy with or without hydroxyurea: ECLAP follow-up data. Blood Cancer J. 2018, 8, 5.
- Hasselbalch, H.C. Smoking as a contributing factor for development of polycythemia vera and related neoplasms. Leuk Res. 2015, S0145–S2126, 30373–30378.
- Pedersen, K.M.; Bak, M.; Sørensen, A.L.; Zwisler, A.D.; Ellervik, C.; Larsen, M.K.; Hasselbalch, H.C.; Tolstrup, J.S. Smoking is associated with increased risk of myeloproliferative neoplasms: A general population-based cohort study. Cancer Med. 2018, 7, 5796–5802.
- Hasselbalch, H.C. Perspectives on chronic inflammation in essential thrombocythemia, polycythemia vera, and myelofibrosis: Is chronic inflammation a trigger and driver of clonal evolution and development of accelerated atherosclerosis and second cancer? Blood 2012, 119, 3219–3225.
- Zhang, C. The role of inflammatory cytokines in endothelial dysfunction. Basic Res. Cardiol. 2008, 103, 398–406.
- Folsom, A.R.; Lutsey, P.L.; Astor, B.C.; Cushman, M. C-reactive protein and venous thromboembolism. A prospective investigation in the ARIC cohort. Thromb Haemost 2009, 102, 615–619.
- Di Battista, V.; Bochicchio, M.T.; Giordano, G.; Napolitano, M.; Lucchesi, A. Genetics and Pathogenetic Role of Inflammasomes in Philadelphia Negative Chronic Myeloproliferative Neoplasms: A Narrative Review. Int. J. Mol. Sci. 2021, 22, 561.
- Jelinek, J.; Oki, Y.; Gharibyan, V.; Bueso-Ramos, C.; Prchal, J.T.; Verstovsek, S.; Beran, M.; Estey, E.; Kantarjian, H.M.; Issa, J.P.J. JAK2 mutation 1849G > T is rare in acute leukemias but can be found in CMML, Philadelphia chromosome-negative CML, and megakaryocytic leukemia. Blood 2005, 106, 3370–3373.
- Levine, R.L.; Loriaux, M.; Huntly, B.J.; Loh, M.L.; Beran, M.; Stoffregen, E.; Berger, R.; Clark, J.J.; Willis, S.G.; Nguyen, K.T.; et al. The JAK2 V617F activating mutation occurs in chronic myelomonocytic leukemia and acute myeloid leukemia, but not in acute lymphoblastic leukemia or chronic lymphocytic leukemia. Blood 2005, 106, 3377–3379.
- Sidon, P.; El Housni, H.; Dessars, B.; Heimann, P. The JAK2V617F mutation is detectable at very low level in peripheral blood of healthy donors. Leukemia 2006, 20, 1622.
- Klippel, S.; Strunck, E.; Temerinac, S.; Bench, A.J.; Meinhardt, G.; Mohr, U.; Leichtle, R.; Green, A.R.; Griesshammer, M.; Heimpel, H.; et al. Quantification of PRV-1 mRNA distinguishes polycythemia vera from secondary erythrocytosis. Blood 2003, 102, 3569–3574.
- Martini, M.; Teofili, L.; Larocca, L.M. Overexpression of PRV-1 gene in polycythemia rubra vera and essential thrombocythemia. Methods Mol. Med. 2006, 125, 265–273.
- Bock, O.; Serinsöz, E.; Neusch, M.; Schlué, J.; Kreipe, H. The polycythaemia rubra vera-1 gene is constitutively expressed by bone marrow cells and does not discriminate polycythaemia vera from reactive and other chronic myeloproliferative disorders. Br. J. Haematol. 2003, 123, 472–474.
- Mirza, A.M.; Correa, P.N.; Axelrad, A.A. Increased basal and induced tyrosine phosphorylation of the insulin-like growth factor I receptor beta subunit in circulating mononuclear cells of patients with polycythemia vera. Blood 1995, 86, 877–882.
- Wang, J.C.; Shi, G.; Baptiste, S.; Yarotska, M.; Sindhu, H.; Wong, C.; Kalavar, M.; Gotlieb, V.; Bandarchuk, A.; Chen, H. Quantification of IGF-1 Receptor May Be Useful in Diagnosing Polycythemia Vera-Suggestion to Be Added to Be One of the Minor Criterion. PLoS ONE 2016, 11, e0165299.
- Johansson, P.L.; Safai-Kutti, S.; Kutti, J. An elevated venous haemoglobin concentration cannot be used as a surrogate marker for absolute erythrocytosis: A study of patients with polycythaemia vera and apparent polycythaemia. Br. J. Haematol. 2005, 129, 701–705.
- Alvarez-Larrán, A.; Ancochea, A.; Angona, A.; Pedro, C.; García-Pallarols, F.; Martínez-Avilés, L.; Bellosillo, B.; Besses, C. Red cell mass measurement in patients with clinically suspected diagnosis of polycythemia vera or essential thrombocythemia. Haematologica 2012, 97, 1704–1707.
- Barbui, T.; Thiele, J.; Carobbio, A. Masked polycythemia vera diagnosed according to WHO and BCSH classification. Am. J. Hematol. 2014, 89, 199–202.
- Barbui, T.; Thiele, J.; Carobbio, A.; Guglielmelli, P.; Rambaldi, A.; Vannucchi, A.M.; Tefferi, A. Discriminating between essential thrombocythemia and masked polycythemia vera in JAK2 mutated patients. Am. J. Hematol. 2014, 89, 588–590.
- Silver, R.T.; Chow, W.; Orazi, A.; Arles, S.P.; Goldsmith, S.J. Evaluation of WHO criteria for diagnosis of polycythemia vera: A prospective analysis. Blood 2013, 122, 1881–1886.
- Barbui, T.; Thiele, J.; Gisslinger, H.; Finazzi, G.; Carobbio, A.; Rumi, E.; Luigia Randi, M.; Bertozzi, I.; Vannucchi, A.M.; Pieri, L.; et al. Masked polycythemia vera (mPV): Results of an international study. Am. J. Hematol. 2014, 89, 52–54.
- Lussana, F.; Carobbio, A.; Randi, M.L.; Elena, C.; Rumi, E.; Finazzi, G.; Bertozzi, I.; Pieri, L.; Ruggeri, M.; Palandri, F.; et al. A lower intensity of treatment may underlie the increased risk of thrombosis in young patients with masked polycythaemia vera. Br. J. Haematol. 2014, 167, 541–546.
- Song, J.; Hussaini, M.; Zhang, H.; <named-content content-type="background:white">Shao, H.; Qin, D.; Zhang, X.; Ma, Z.; Hussnain Naqvi, S.M.; Zhang, L.; Moscinski, L.C. Comparison of the mutational profiles of Primary Myelofibrosis, Polycythemia Vera, and Essential Thrombocytosis. Am. J. Clin. Pathol. 2017, 147, 444–452.
- Goerttler, P.S.; Kreutz, C.; Donauer, J.; Faller, D.; Maiwald, T.; März, E.; Rumberger, B.; Sparna, T.; Schmitt-Gräff, A.; Wilpert, J.; et al. Gene expression profiling in polycythaemia vera: Overexpression of transcription factor NF-E2. Br. J. Haematol. 2005, 129, 138–150.
- Gallardo, M.; Barrio, S.; Fernandez, M.; Paradela, A.; Arenas, A.; Toldos, O.; Ayala, R.; Albizua, E.; Jimenez, A.; Redondo, S.; et al. Proteomic analysis reveals heat shock protein 70 has a key role in polycythemia Vera. Mol. Cancer 2013, 12, 142.
- Brusson, M.; Cochet, S.; Leduc, M.; Guillonneau, F.; Mayeux, P.; Peyrard, T.; Chomienne, C.; Le Van Kim, C.; Cassinat, B.; Kiladjian, J.J.; et al. Enhanced calreticulin expression in red cells of polycythemia vera patients harboring the JAK2V617F mutation. Haematologica 2017, 102, e241–e244.
- Bruchova, H.; Merkerova, M.; Prchal, J.T. Aberrant expression of microRNA in polycythemia vera. Haematologica 2008, 93, 1009–1016.
- Zhan, H.; Cardozo, C.; Yu, W.; Wang, A.; Moliterno, A.R.; Dang, C.V.; Spivak, J.L. MicroRNA deregulation in polycythemia vera and essential thrombocythemia patients. Blood Cells Mol. Dis. 2013, 50, 190–195.
- Campbell, P.J.; Scott, L.M.; Buck, G.; Wheatley, K.; East, C.L.; Marsden, J.T.; Duffy, A.; Boyd, E.M.; Bench, A.J.; Scott, M.A.; et al. Definition of subtypes of essential thrombocythaemia and relation to polycythaemia vera based on JAK2 V617F mutation status: A prospective study. Lancet 2005, 366, 1945–1953.
- Passamonti, F.; Rumi, E.; Pietra, D.; Della Porta, M.G.; Boveri, E.; Pascutto, C.; Vanelli, L.; Arcaini, L.; Burcheri, S.; Malcovati, L.; et al. Relation between JAK2 (V617F) mutation status, granulocyte activation, and constitutive mobilization of CD34+ cells into peripheral blood in myeloproliferative disorders. Blood 2006, 107, 3676–3682.
- Alshemmari, S.H.; Rajaan, R.; Ameen, R.; Al-Drees, M.A.; Almosailleakh, M.R. JAK2V617F allele burden in patients with myeloproliferative neoplasms. Ann. Hematol. 2014, 93, 791–796.
- Grinfeld, J.; Nangalia, J.; Baxter, E.J.; Wedge, D.C.; Angelopoulos, N.; Cantrill, R.; Godfrey, A.L.; Papaemmanuil, E.; Gundem, G.; MacLean, C.; et al. Classification and Personalized Prognosis in Myeloproliferative Neoplasms. N. Engl. J. Med. 2018, 379, 1416–1430.
- Tiedt, R.; Hao-Shen, H.; Sobas, M.A.; Looser, R.; Dirnhofer, S.; Schwaller, J.; Skoda, R.C. Ratio of mutant JAK2-V617F to wild-type Jak2 determines the MPD phenotypes in transgenic mice. Blood 2008, 111, 3931–3940.
- Li, J.; Kent, D.G.; Godfrey, A.L.; Manning, H.; Nangalia, J.; Aziz, A.; Chen, E.; Saeb-Parsy, K.; Fink, J.; Sneade, R.; et al. JAK2V617FJAK2V617F homozygosity drives a phenotypic switch between myeloproliferative neoplasms in a murine model, but is insufficient to sustain disease. Blood 2014, 123, 3139–3151.
- Chaiter, Y.; Brenner, B.; Aghai, E.; Tatarsky, I. High incidence of myeloproliferative disorders in Ashkenazi Jews in northern Israel. Leuk Lymphoma 1992, 7, 251–255.
- Landgren, O.; Goldin, L.R.; Kristinsson, S.Y.; Helgadottir, E.A.; Samuelsson, J.; Bjorkholm, M. Increased risks of polycythemia vera, essential thrombocythemia, and myelofibrosis among 24577 first-degree relatives of 11039 patients with myeloproliferative neoplasms in Sweden. Blood 2008, 112, 2199–2204.
- Rumi, E.; Passamonti, F.; Della Porta, M.G.; Elena, C.; Arcaini, L.; Vanelli, L.; Del Curto, C.; Pietra, D.; Boveri, E.; Pascutto, C.; et al. Familial chronic myeloproliferative disorders: Clinical phenotype and evidence of disease anticipation. J. Clin. Oncol. 2007, 25, 5630–5635.
- Jones, A.V.; Chase, A.; Silver, R.T.; Oscier, D.; Zoi, K.; Wang, Y.L.; Cario, H.; Pahl, H.L.; Collins, A.; Reiter, A.; et al. JAK2 haplotype is a major risk factor for the development of myeloproliferative neoplasms. Nat. Genet. 2009, 41, 446–449.
- Olcaydu, D.; Harutyunyan, A.; Jäger, R.; Berg, T.; Gisslinger, B.; Pabinger, I.; Gisslinger, H.; Kralovics, R. A common JAK2 haplotype confers susceptibility to myeloproliferative neoplasms. Nat. Genet. 2009, 41, 450–454.
- Kilpivaara, O.; Mukherjee, S.; Schram, A.M.; Wadleigh, M.; Mullally, A.; Ebert, B.L.; Bass, A.; Marubayashi, S.; Heguy, A.; Garcia-Manero, G.; et al. A germline JAK2 SNP is associated with predisposition to the development of JAK2(V617F)- positive myeloproliferative neoplasms. Nat. Genet. 2009, 41, 455–459.
- Olcaydu, D.; Skoda, R.C.; Looser, R.; Li, S.; Cazzola, M.; Pietra, D.; Passamonti, F.; Lippert, E.; Carillo, S.; Girodon, F.; et al. The ‘GGCC’ haplotype of JAK2 confers susceptibility to JAK2 exon 12 mutation-positive polycythemia vera. Leukemia 2009, 23, 1924–1926.
- Nussenzveig, R.H.; Swierczek, S.I.; Jelinek, J.; Gaikwad, A.; Liu, E.; Verstovsek, S.; Prchal, J.F.; Prchal, J.T. Polycythemia vera is not initiated by JAK2V617F mutation. Exp. Hematol. 2007, 35, 32–38.
- Kralovics, R.; Teo, S.S.; Li, S.; Theocharides, A.; Buser, A.S.; Tichelli, A.; Skoda, R.C. Acquisition of the V617F mutation of JAK2 is a late genetic event in a subset of patients with myeloproliferative disorders. Blood 2006, 108, 1377–1380.
- Rumi, E.; Passamonti, F.; Pietra, D.; Della Porta, M.G.; Arcaini, L.; Boggi, S.; Elena, C.; Boveri, E.; Pascutto, C.; Lazzarino, M.; et al. JAK2 (V617F) as an acquired somatic mutation and a secondary genetic event associated with disease progression in familial myeloproliferative disorders. Cancer 2006, 107, 2206–2211.
- James, C. The JAK2V617F Mutation in Polycythemia Vera and Other Myeloproliferative Disorders: One Mutation for Three Diseases? Hematology Am. Soc. Hematol. Educ. Program. 2008, 2008, 69–75.
- Lanikova, L.; Babosova, O.; Swierczek, S.; Wang, L.; Wheeler, D.A.; Divoky, V.; Korinek, V.; Prchal, J.T. Coexistence of gain-of-function JAK2 germ line mutations with JAK2V617F in polycythemia vera. Blood 2016, 128, 2266–2270.
- Harutyunyan, A.S.; Giambruno, R.; Krendl, C.; Stukalov, A.; Klampfl, T.; Berg, T.; Milosevic, J.D.; Chen, D.; Gisslinger, B.; Gisslinger, H.; et al. Germline RBBP6 Mutations In Myeloproliferative Neoplasms [abstract]. Blood 2013, 122, 267.
- Oddsson, A.; Kristinsson, S.Y.; Helgason, H.; Gudbjartsson, D.F.; Masson, G.; Sigurdsson, A.; Jonasdottir, A.; Jonasdottir, A.; Steingrimsdottir, H.; Vidarsson, B.; et al. The germline sequence variant rs2736100_C in TERT associates with myeloproliferative neoplasms. Leukemia 2014, 28, 1371–1374.
- Harutyunyan, A.S.; Giambruno, R.; Krendl, C.; Stukalov, A.; Klampfl, T.; Berg, T.; Chen, D.; Milosevic Feenstra, J.D.; Jäger, R.; Gisslinger, B.; et al. Germline RBBP6 mutations in familial myeloproliferative neoplasms. Blood 2016, 127, 362–365.
- Bao, E.L.; Nandakumar, S.K.; Liao, X.; Bick, A.G.; Karjalainen, J.; Tabaka, M.; Gan, O.I.; Havulinna, A.S.; Kiiskinen, T.; Lareau, C.A.; et al. Inherited myeloproliferative neoplasm risk affects haematopoietic stem cells. Nature 2020, 586, 769–775.
- Giaccherini, M.; Macauda, A.; Sgherza, N.; Sainz, J.; Gemignani, F.; Maldonado, J.; Jurado, M.; Tavano, F.; Mazur, G.; Jerez, A.; et al. Genetic polymorphisms associated with telomere length and risk of developing myeloproliferative neoplasms. Blood Cancer J. 2020, 10, 89.
- Williams, N.; Lee, J.; Moore, L.; Baxter, J.E.; Hewinson, J.; Dawson, K.J.; Menzies, A.; Godfrey, A.L.; Green, A.R.; Campbell, P.J.; et al. Driver mutation acquisition in utero and childhood followed by lifelong clonal evolution underlie myeloproliferative neoplasms. 2020 ASH Annual Meeting & Exposition. Abstract LBA-1. Presented December 8, 2020. Available online: (accessed on 15 February 2021).
- Nangalia, J.; Green, T.R. The evolving genomic landscape of myeloproliferative neoplasms. Hematology Am. Soc. Hematol. Educ. Program. 2014, 2014, 287–296.
- Tefferi, A.; Lasho, T.L.; Guglielmelli, P.; Finke, C.M.; Rotunno, G.; Elala, Y.; Pacilli, A.; Hanson, C.A.; Pancrazzi, A.; Ketterling, R.P.; et al. Targeted deep sequencing in polycythemia vera and essential thrombocythemia. Blood Adv. 2016, 1, 21–30.
- Segura-Díaz, A.; Stuckey, R.; Florido, Y.; González-Martín, J.M.; López-Rodríguez, J.F.; Sánchez-Sosa, S.; González-Pérez, E.; Sáez Perdomo, M.N.; Perera, M.; de la Iglesia, S.; et al. Thrombotic Risk Detection in Patients with Polycythemia Vera: The Predictive Role of DNMT3A/TET2/ASXL1 Mutations. Cancers 2020, 12, 934.
- Green, A.; Beer, P. Somatic mutations of IDH1 and IDH2 in the leukemic transformation of myeloproliferative neoplasms. N. Engl. J. Med. 2010, 362, 369–370.
- Figueroa, M.E.; Abdel-Wahab, O.; Lu, C.; Ward, P.S.; Patel, J.; Shih, A.; Li, Y.; Bhagwat, N.; Vasanthakumar, A.; Fernandez, H.F.; et al. Leukemic IDH1 and IDH2 mutations result in a hypermethylation phenotype, disrupt TET2 function, and impair hematopoietic differentiation. Cancer Cell 2010, 18, 553–567.
- Marcault, C.; Zhao, L.P.; Daltro De Oliveira, R.; Soret, J.; Gauthier, N.; Verger, E.; Maslah, N.; Roux, B.; Parquet, N.; Dosquet, C.; et al. NFE2 Mutations Impact AML Transformation and Overall Survival in Patients with Myeloproliferative Neoplasms (MPN). Blood 2020, 136 (Suppl. 1), 36.
- Marinaccio, C.; Suraneni, P.K.; Celik, H.; Volk, A.; Wen, J.Q.; Ling, T.; Lasho, T.; Koche, R.P.; Famulare, C.; Stein, B.L.; et al. Loss of LKB1/STK11 Facilitates Leukemic Progression of the Myeloproliferative Neoplasms. Blood 2020, 136 (Suppl. 1), 1.
- Laurie, C.C.; Laurie, C.A.; Rice, K.; Doheny, K.F.; Zelnick, L.R.; McHugh, C.P.; Ling, H.; Hetrick, K.N.; Pugh, E.W.; Amos, C.; et al. Detectable clonal mosaicism from birth to old age and its relationship to cancer. Nat. Genet. 2012, 44, 642–650.
- Busque, L.; Patel, J.P.; Figueroa, M.E.; Vasanthakumar, A.; Provost, S.; Hamilou, Z.; Mollica, L.; Li, J.; Viale, A.; Heguy, A.; et al. Recurrent somatic TET2 mutations in normal elderly individuals with clonal hematopoiesis. Nat. Genet. 2012, 44, 1179–1181.
- Jaiswal, S.; Fontanillas, P.; Flannick, J.; Manning, A.; Grauman, P.V.; Mar, B.G.; Lindsley, R.C.; Mermel, C.H.; Burtt, N.; Chavezet, A.; et al. Age-related clonal hematopoiesis associated with adverse outcomes. N. Engl. J. Med. 2014, 371, 2488–2498.
- Genovese, G.; Kähler, A.K.; Handsaker, R.E.; Lindberg, J.; Rose, S.A.; Bakhoum, S.F.; Chambert, K.; Mick, E.; Neale, B.M.; Fromer, M.; et al. Clonal hematopoiesis and blood-cancer risk inferred from blood DNA sequence. N. Engl. J. Med. 2014, 371, 2477–2487.
- Xie, M.; Lu, C.; Wang, J.; McLellan, M.D.; Johnson, K.J.; Wendl, M.C.; McMichael, J.F.; Schmidt, H.K.; Yellapantula, V.; Miller, C.A.; et al. Age-related mutations associated with clonal hematopoietic expansion and malignancies. Nat. Med. 2014, 20, 1472–1478.
- Steensma, D.P.; Bejar, R.; Jaiswal, S.; Lindsley, C.; Sekeres, M.A.; Hasserjian, R.P.; Ebert, B.L. Clonal hematopoiesis of indeterminate potential and its distinction from myelodysplastic syndromes. Blood 2015, 126, 9–16.
- Jaiswal, S.; Natarajan, P.; Silver, A.J.; Gibson, C.J.; Bick, A.G.; Shvartz, E.; McConkey, M.; Gupta, N.; Gabriel, S.; Ardissino, D.; et al. Clonal hematopoiesis and risk of atherosclerotic cardiovascular disease. N. Engl. J. Med. 2017, 377, 111–121.
- Tefferi, A.; Pardanani, A.; Lim, K.H.; Abdel-Wahab, O.; Lasho, T.L.; Patel, J.; Gangat, N.; Finke, C.M.; Schwager, S.; Mullally, A.; et al. TET2 mutations and their clinical correlates in polycythemia vera, essential thrombocythemia and myelofibrosis. Leukemia 2009, 23, 905–911.
- Delhommeau, F.; Dupont, S.; Della Valle, V.; James, C.; Trannoy, S.; Massé, A.; Kosmider, O.; Le Couedic, J.P.; Robert, F.; Alberdi, A.; et al. Mutation in TET2 in myeloid cancers. N. Engl. J. Med. 2009, 360, 2289–2301.
- Kameda, T.; Shide, K.; Yamaji, T.; Kamiunten, A.; Sekine, M.; Taniguchi, Y.; Hidaka, T.; Kubuki, Y.; Shimoda, H.; Marutsuka, K.; et al. Loss of TET2 has dual roles in murine myeloproliferative neoplasms: Disease sustainer and disease accelerator. Blood 2015, 125, 304–315.
- Chen, E.; Schneider, R.K.; Breyfogle, L.J.; Rosen, E.A.; Poveromo, L.; Elf, S.; Ko, A.; Brumme, K.; Levine, R.; Ebert, B.L.; et al. Distinct effects of concomitant Jak2V617F expression and Tet2 loss in mice promote disease progression in myeloproliferative neoplasms. Blood 2015, 125, 327–335.
- Hughes, A.E.; Magrini, V.; Demeter, R.; Miller, C.A.; Fulton, R.; Fulton, L.L.; Eades, W.C.; Elliott, K.; Heath, S.; Westervelt, P.; et al. Clonal architecture of secondary acute myeloid leukemia defined by single-cell sequencing. PLoS Genet. 2014, 10, e1004462.
- Nice, F.L.; Massie, C.E.; Klampfl, T.; Green, A.R. Determination of complex subclonal structures of hematological malignancies by multiplexed genotyping of blood progenitor colonies. Exp. Hematol. 2018, 57, 60–64.
- Yang, L.; Rau, R.; Goodell, M.A. DNMT3A in haematological malignancies. Nat. Rev. Cancer 2015, 15, 152–165.
- Ortmann, C.A.; Kent, D.G.; Nangalia, J.; Silber, Y.; Wedge, D.C.; Grinfeld, J.; Baxter, E.J.; Massie, C.E.; Papaemmanuil, E.; Menon, S.; et al. Effect of mutation order on myeloproliferative neoplasms. N. Engl. J. Med. 2015, 372, 601–612.
- Tefferi, A.; Guglielmelli, P.; Larson, D.R.; Finke, C.; Wassie, E.A.; Pieri, L.; Gangat, N.; Fjerza, R.; Belachew, A.A.; Lasho, T.L.; et al. Long-term survival and blast transformation in molecularly annotated essential thrombocythemia, polycythemia vera, and myelofibrosis. Blood 2014, 124, 2507–2513.
- McMullin, M.F.; Bareford, D.; Campbell, P.; Green, A.R.; Harrison, C.; Hunt, B.; Oscier, D.; Polkey, M.I.; Reilly, J.T.; Rosenthal, E.; et al. Guidelines for the diagnosis, investigation and management of polycythaemia/erythrocytosis. Br. J. Haematol. 2005, 130, 174–195.
- Tefferi, A.; Guglielmelli, P.; Lasho, T.L.; Coltro, G.; Finke, C.M.; Loscocco, G.G.; Sordi, B.; Szuber, N.; Rotunno, G.; Pacilli, A.; et al. Mutation-enhanced international prognostic systems for essential thrombocythaemia and polycythaemia vera. Br. J. Haematol. 2020, 189, 291–302.
- Vannucchi, A.M.; Antonioli, E.; Guglielmelli, P.; Rambaldi, A.; Barosi, G.; Marchioli, R.; Marfisi, R.M.; Finazzi, G.; Guerini, V.; Fabris, F.; et al. Clinical profile of homozygous JAK2 617V>F mutation in patients with polycythemia vera or essential thrombocythemia. Blood 2007, 110, 840–846.
- Vannucchi, A.M.; Antonioli, E.; Guglielmelli, P.; Longo, G.; Pancrazzi, A.; Ponziani, V.; Bogani, C.; Ferrini, P.R.; Rambaldi, A.; Guerini, V.; et al. MPD Research Consortium. Prospective identification of high-risk polycythemia vera patients based on JAK2(V617F) allele burden. Leukemia 2007, 21, 1952–1959.
- Gangat, N.; Strand, J.J.; Lasho, T.L.; Li, C.Y.; Pardanani, A.; Tefferi, A. Pruritus in polycythemia vera is associated with a lower risk of arterial thrombosis. Am. J. Hematol. 2008, 83, 451–453.
- Larsen, T.S.; Pallisgaard, N.; Møller, M.B.; Hasselbalch, H.C. The JAK2 V617F allele burden in essential thrombocythemia, polycythemia vera and primary myelofibrosis--impact on disease phenotype. Eur. J. Haematol. 2007, 79, 508–515.
- Kirschner, M.M.; Schemionek, M.; Schubert, C.; Chatain, N.; Sontag, S.; Isfort, S.; Ortiz-Brüchle, N.; Schmitt, K.; Krüger, L.; Zerres, K.; et al. Dissecting Genomic Aberrations in Myeloproliferative Neoplasms by Multiplex-PCR and Next Generation Sequencing. PLoS ONE 2015, 10, e0123476.
- Borowczyk, M.; Wojtaszewska, M.; Lewandowski, K.; Gil, L.; Lewandowska, M.; Lehmann-Kopydłowska, A.; Kroll-Balcerzak, R.; Balcerzak, A.; Iwoła, M.; Michalak, M.; et al. The JAK2 V617F mutational status and allele burden may be related with the risk of venous thromboembolic events in patients with Philadelphia-negative myeloproliferative neoplasms. Thromb Res. 2015, 135, 272–280.
- Malak, S.; Labopin, M.; Saint-Martin, C.; Bellanne-Chantelot, C.; Najman, A.; French Group of Familial Myeloproliferative Disorders. Long term follow up of 93 families with myeloproliferative neoplasms: Life expectancy and implications of JAK2V617F in the occurrence of complications. Blood Cells Mol. Dis. 2012, 49, 170–176.
- Kralovics, R.; Teo, S.S.; Buser, A.S.; Brutsche, M.; Tiedt, R.; Tichelli, A.; Passamonti, F.; Pietra, D.; Cazzola, M.; Skoda, R.C. Altered gene expression in myeloproliferative disorders correlates with activation of signaling by the V617F mutation of Jak2. Blood 2005, 106, 3374–3376.
- Vainchenker, W.; Constantinescu, S.N. A Unique Activating Mutation in JAK2 (V617F) Is at the Origin of Polycythemia Vera and Allows a New Classification of Myeloproliferative Diseases. Hematology Am. Soc. Hematol. Educ. Program. 2005, 195–200.
- Vannucchi, A.M.; Antonioli, E.; Guglielmelli, P.; Pardanani, A.; Tefferi, A. Clinical correlates of JAK2V617F presence or allele burden in myeloproliferative neoplasms: A critical reappraisal. Leukemia 2008, 22, 1299–1307.
- Vannucchi, A.M.; Antonioli, E.; Guglielmeli, P.; Pancrazzi, A.; Bogani, C.; Pieri, L.; Bosi, A. Influence of the JAK2V617F mutational load at diagnosis on major clinical aspects in patients with Polycythemia vera. Blood 2006, 108, 5.
- Vannucchi, A.M.; Barosi, G.; Rimbaldi, A.; Barosi, G.; Rambaldi, A.; Marchioli, R.; Barbui, T.; Gimema, W.P. Clinical significance of JAK2V617F homozygosity in the chronic myeloproliferative disorders. A study of 1306 patients. Blood 2006, 108, 664.
- Spivak, J.L.; Considine, M.; Williams, D.M.; Talbot, C.C., Jr.; Rogers, O.; Moliterno, A.R.; Jie, C.; Ochs, M.F. Two clinical phenotypes in polycythemia vera. N. Engl. J. Med. 2014, 371, 808–817.
- Swolin, B.; Weinfeld, A.; Westin, J. A prospective long-term cytogenetic study in polycythemia vera in relation to treatment and clinical course. Blood 1988, 72, 386–395.
- Diez-Martin, J.L.; Graham, D.L.; Petitt, R.M.; Dewald, G.W. Chromosome studies in 104 patients with polycythemia vera. Mayo Clin. Proc. 1991, 66, 287–299.
- Gangat, N.; Strand, J.; Lasho, T.L.; Finke, C.M.; Knudson, R.A.; Pardanani, A.; Li, C.Y.; Ketterling, R.P.; Tefferi, A. Cytogenetic studies at diagnosis in polycythemia vera: Clinical and JAK2V617F allele burden correlates. Eur. J. Haematol. 2008, 80, 197–200.
- Finazzi, F.; Caruso, V.; Marchioli, R.; Capnist, G.; Chisesi, T.; Finelli, C.; Gugliotta, L.; Landolfi, R.; Kutti, J.; Gisslinger, H.; et al. Acute leukemia in polycythemia vera: An analysis of 1638 patients enrolled in a prospective observational study. Blood 2005, 105, 2664–2670.
- Tang, G.; Hidalgo Lopez, J.E.; Wang, S.A.; Hu, S.; Ma, J.; Pierce, S.; Zuo, W.; Carballo-Zarate, A.A.; Yin, C.C.; Tang, Z.; et al. Characteristics and clinical significance of cytogenetic abnormalities in polycythemia vera. Haematologica 2017, 102, 1511–1518.
- Sever, M.; Quintás-Cardama, A.; Pierce, S.; Zhou, L.; Kantarjian, H.; Verstovsek, S. Significance of cytogenetic abnormalities in patients with polycythemia vera. Leuk Lymphoma 2013, 54, 2667–2670.
- Tefferi, A.; Rumi, E.; Finazzi, G.; Gisslinger, H.; Vannucchi, A.M.; Rodeghiero, F.; Randi, M.L.; Vaidya, R.; Cazzola, M.; Rambaldi, A.; et al. Survival and prognosis among 1545 patients with contemporary polycythemia vera: An international study. Leukemia 2013, 27, 1874–1881.
- Cerquozzi, S.; Tefferi, A. Blast transformation and fibrotic progression in polycythemia vera and essential thrombocythemia: A literature review of incidence and risk factors. Blood Cancer J. 2015, 5, e366.
- Barbui, T.; Vannucchi, A.M.; Carobbio, A.; Thiele, J.; Rumi, E.; Gisslinger, H.; Rodeghiero, F.; Randi, M.L.; Rambaldi, A.; Pieri, L.; et al. Patterns of presentation and thrombosis outcome in patients with polycythemia vera strictly defined by WHO-criteria and stratified by calendar period of diagnosis. Am. J. Hematol. 2015, 90, 434–437.
- Barbui, T.; Barosi, G.; Birgegard, G.; Cervantes, F.; Finazzi, G.; Griesshammer, M.; Harrison, C.; Hasselbalch, H.C.; Hehlmann, R.; Hoffman, R.; et al. Philadelphia-negative classical myeloproliferative neoplasms: Critical concepts and management recommendations from European LeukemiaNet. J. Clin. Oncol. 2011, 29, 761–770.
- Vannucchi, A.M. JAK2 mutation and thrombosis in the myeloproliferative neoplasms. Curr. Hematol. Malig Rep. 2010, 5, 22–28.
- Chia, Y.C.; Islam, M.A.; Woon, P.Y.; Johan, M.F.; Hassan, R.; Ramli, M. Molecular genetics of thrombotic myeloproliferative neoplasms: Implications in precision oncology. Genes Dis. 2021.
- Ruggeri, M.; Gisslinger, H.; Tosetto, A.; Rintelen, C.; Mannhalter, C.; Pabinger, I.; Heis, N.; Castaman, G.; Missiaglia, E.; Lechner, K.; et al. Leiden mutation carriership and venous thromboembolism in polycythemia vera and essential thrombocythemia. Am. J. Hematol. 2002, 71, 1–6.
- Jensen, M.K.; de Nully Brown, P.; Thorsen, S.; Hasselbalch, H.C. Frequent occurrence of anticardiolipin antibodies, Factor V Leiden mutation, and perturbed endothelial function in chronic myeloproliferative disorders. Am. J. Hematol. 2002, 69, 185–191.
- Trifa, A.P.; Cucuianu, A.; Popp, R.A.; Coadă, C.A.; Costache, R.M.; Militaru, M.S.; Vesa, S.C.; Pop, I.V. The relationship between factor V Leiden, prothrombin G20210A, and MTHFR mutations and the first major thrombotic episode in polycythemia vera and essential thrombocythemia. Ann. Hematol. 2014, 93, 203–209.
- Algahtani, F.H.; Stuckey, R. High factor VIII levels and arterial thrombosis: Illustrative case and literature review. Ther. Adv. Hematol. 2019, 10, 2040620719886685.
- Sacco, M.; Ranalli, P.; Lancellotti, S.; Petrucci, G.; Dragani, A.; Rocca, B.; De Cristofaro, R. Increased von Willebrand factor levels in polycythemia vera and phenotypic differences with essential thrombocythemia. Res. Pract Thromb Haemost. 2020, 4, 413–421.
- De Stefano, V.; Za, T.; Rossi, E.; Vannucchi, A.M.; Ruggeri, M.; Elli, E.; Micò, C.; Tieghi, A.; Cacciola, R.R.; Santoro, C.; et al. Leukocytosis is a risk factor for recurrent arterial thrombosis in young patients with polycythemia vera and essential thrombocythemia. Am. J. Hematol. 2010, 85, 97–100.
- Carobbio, A.; Ferrari, A.; Masciulli, A.; Ghirardi, A.; Barosi, G.; Barbui, T. Leukocytosis and thrombosis in essential thrombocythemia and polycythemia vera: A systematic review and meta-analysis. Blood Adv. 2019, 3, 1729–1737.
- Barbui, T.; Finazzi, G.; Carobbio, A.; Thiele, J.; Passamonti, F.; Rumi, E.; Ruggeri, M.; Rodeghiero, F.; Randi, M.L.; Bertozzi, I.; et al. Development and validation of an International Prognostic Score of thrombosis in World Health Organization-essential thrombocythemia (IPSET-thrombosis). Blood 2012, 120, 5128–5133.
- Carobbio, A.; Finazzi, G.; Antonioli, E.; Guglielmelli, P.; Vannucchi, A.M.; Dellacasa, C.M.; Salmoiraghi, S.; Delaini, F.; Rambaldi, A.; Barbui, T. JAK2V617F allele burden and thrombosis: A direct comparison in essential thrombocythemia and polycythemia vera. Exp. Hematol. 2009, 37, 1016–1021.
- Horvat, I.; Boban, A.; Zadro, R.; Antolic, M.R.; Serventi-Seiwerth, R.; Roncevic, P.; Radman, I.; Sertic, D.; Vodanovic, M.; Pulanic, D.; et al. Influence of Blood Count, Cardiovascular Risks, Inherited Thrombophilia, and JAK2 V617F Burden Allele on Type of Thrombosis in Patients with Philadelphia Chromosome Negative Myeloproliferative Neoplasms. Clin. Lymphoma Myeloma Leuk 2019, 19, 53–63.
- Barbui, T.; Carobbio, A.; Finazzi, G.; Vannucchi, A.M.; Barosi, G.; Antonioli, E.; Guglielmelli, P.; Pancrazzi, A.; Salmoiraghi, S.; Zilio, P.; et al. Inflammation and thrombosis in essential thrombocythemia and polycythemia vera: Different role of C-reactive protein and pentraxin 3. Haematologica 2011, 96, 315–318.
- Fuster, J.J.; MacLauchlan, S. Clonal hematopoiesis associated with TET2 deficiency accelerates atherosclerosis development in mice. Science 2017, 355, 842–847.
- Ridker, P.M.; MacFadyen, J.G.; Thuren, T.; Libby, P. Residual inflammatory risk associated with interleukin-18 and interleukin-6 after successful interleukin-1b inhibition with canakinumab: Further rationale for the development of targeted anti-cytokine therapies for the treatment of atherothrombosis. Eur. Heart J. 2019, 41, 2153–2163.
- CARDIoGRAMplusC4D Consortium; Deloukas, P.; Kanoni, S.; Willenborg, C.; Farrall, M.; Assimes, T.L.; Thompson, J.R.; Ingelsson, E.; Saleheen, D.; Erdmann, J.; et al. Large--scale association analysis identifies new risk loci for coronary artery disease. Nat. Genet. 2013, 45, 25–33.
- Wang, W.; Tang, Y.; Wang, Y.; Tascau, L.; Balcerek, J.; Tong, W.; Levine, R.L.; Welch, C.; Tall, A.R.; Wang, N. LNK/SH2B3 Loss of Function Promotes Atherosclerosis and Thrombosis. Circ Res. 2016, 119, e91–e103.
- Kaplar, J.K.; Kiss, A.K.; Szabo, M.; Udvardy, M. Increased leukocyte-platelet adhesion in chronic myeloproliferative disorders with high platelet counts. Platelets 2000, 11, 183–184.
- Jensen, M.K.; de Nully Brown, P.; Lund, B.V.; Nielsen, O.J.; Hasselbalch, H.C. Increased circulating platelet–leukocyte aggregates in myeloproliferative disorders is correlated to previous thrombosis, platelet activation and platelet count. Eur. J. Haematol. 2001, 66, 143–151.
- Griesshammer, M.; Klippel, S.; Strunck, E.; Temerinac, S.; Mohr, U.; Heimpel, H.; Pahl, H.L. PRV-1 mRNA expression discriminates two types of essential thrombocythemia. Ann. Hematol. 2004, 83, 364–370.
- Shahrabi, S.; Ehsanpour, A.; Heidary, S.; Shahjahani, M.; Behzad, M.M. Expression of CD markers in JAK2V617F positive myeloproliferative neoplasms: Prognostic significance. Oncol. Rev. 2018, 12, 373.
- Ferrer-Marín, F.; Cuenca-Zamora, E.J.; Guijarro-Carrillo, P.J.; Teruel-Montoya, R. Emerging Role of Neutrophils in the Thrombosis of Chronic Myeloproliferative Neoplasms. Int. J. Mol. Sci. 2021, 22, 1143.
- Ma, X.; Vanasse, G.; Cartmel, B.; Wang, Y.; Selinger, H.A. Prevalence of polycythemia vera and essential thrombocythemia. Am. J. Hematol. 2008, 83, 359–362.
- Benton, C.B.; Tanaka, M.; Wilson, C.; Pierce, S.; Zhou, L.; Cortes, J.; Kantarjian, H.; Verstovsek, S. Increased likelihood of post-polycythemia vera myelofibrosis in Ph-negative MPN patients with chromosome 12 abnormalities. Leuk Res. 2015, 39, 419–423.
- Harutyunyan, A.; Klampfl, T.; Cazzola, M.; Kralovics, R. p53 lesions in leukemic transformation. N. Engl. J. Med. 2011, 364, 488–490.
- Rotunno, G.; Pacilli, A.; Artusi, V.; Rumi, E.; Maffioli, M.; Delaini, F.; Brogi, G.; Fanelli, T.; Pancrazzi, A.; Pietra, D.; et al. Epidemiology and clinical relevance of mutations in postpolycythemia vera and postessential thrombocythemia myelofibrosis: A study on 359 patients of the AGIMM group. Am. J. Hematol. 2016, 91, 681–686.
- Vannucchi, A.M.; Lasho, T.L.; Guglielmelli, P.; Biamonte, F.; Pardanani, A.; Pereira, A.; Finke, C.; Score, J.; Gangat, N.; Mannarelli, C.; et al. Mutations and prognosis in primary myelofibrosis. Leukemia 2013, 27, 1861–1869.
- Gowin, K.; Coakley, M.; Kosiorek, H.; Mesa, R. Discrepancies of applying primary myelofibrosis prognostic scores for patients with post polycythemia vera/essential thrombocytosis myelofibrosis. Haematologica 2016, 101, e405–e406.
- Passamonti, F.; Giorgino, T.; Mora, B.; Guglielmelli, P.; Rumi, E.; Maffioli, M.; Rambaldi, A.; Caramella, M.; Komrokji, R.; Gotlib, J.; et al. A clinical-molecular prognostic model to predict survival in patients with post polycythemia vera and post essential thrombocythemia myelofibrosis. Leukemia 2017, 31, 2726–2731.
- Palandri, F.; Palumbo, G.A.; Iurlo, A.; Polverelli, N.; Benevolo, G.; Breccia, M.; Abruzzese, E.; Tiribelli, M.; Bonifacio, M.; Tieghi, A.; et al. Differences in presenting features, outcome and prognostic models in patients with primary myelofibrosis and post-polycythemia vera and/or post-essential thrombocythemia myelofibrosis treated with ruxolitinib. New perspective of the MYSEC-PM in a large multicenter study. Semin Hematol. 2018, 55, 248–255.
- Mora, B.; Giorgino, T.; Guglielmelli, P.; Rumi, E.; Maffioli, M.; Rambaldi, A.; Caramella, M.; Komrokji, R.; Gotlib, J.; Kiladjian, J.J.; et al. Value of cytogenetic abnormalities in post-polycythemia vera and post-essential thrombocythemia myelofibrosis: A study of the MYSEC project. Haematologica 2018, 103, e392–e394.
- Theocharides, A.; Boissinot, M.; Girodon, F.; Garand, R.; Teo, S.S.; Lippert, E.; Talmant, P.; Tichelli, A.; Hermouet, S.; Skoda, R.C. Leukemic blasts in transformed JAK2-V617F-positive myeloproliferative disorders are frequently negative for the JAK2-V617F mutation. Blood 2007, 110, 375–379.
- Beer, P.A.; Delhommeau, F.; LeCouédic, J.P.; Dawson, M.A.; Chen, E.; Bareford, D.; Kusec, R.; McMullin, M.F.; Harrison, C.N.; Vannucchi, A.M.; et al. Two routes to leukemic transformation after a JAK2 mutation-positive myeloproliferative neoplasm. Blood 2010, 115, 2891–2900.
- Lundberg, P.; Karow, A.; Nienhold, R.; Looser, R.; Hao-Shen, H.; Nissen, I.; Girsberger, S.; Lehmann, T.; Passweg, J.; Stern, M.; et al. Clonal evolution and clinical correlates of somatic mutations in myeloproliferative neoplasms. Blood 2014, 123, 2220–2228.
- Luque Paz, D.; Jouanneau-Courville, R.; Riou, J.; Ianotto, J.C.; Boyer, F.; Chauveau, A.; Renard, M.; Chomel, J.C.; Cayssials, E.; Gallego-Hernanz, M.P.; et al. Leukemic evolution of polycythemia vera and essential thrombocythemia: Genomic profiles predict time to transformation. Blood Adv. 2020, 4, 4887–4897.
- Rampal, R.; Ahn, J.; Abdel-Wahab, O.; Nahas, M.; Wang, K.; Lipson, D.; Otto, G.A.; Yelensky, R.; Hricik, T.; McKenney, A.S.; et al. Genomic and functional analysis of leukemic transformation of myeloproliferative neoplasms. Proc. Natl. Acad. Sci. USA 2014, 111, E5401–E5410.
- Senín, A.; Fernández-Rodríguez, C.; Bellosillo, B.; Camacho, L.; Longarón, R.; Angona, A.; Besses, C.; Álvarez-Larrán, A. Non-driver mutations in patients with JAK2V617F-mutated polycythemia vera or essential thrombocythemia with long-term molecular follow-up. Ann. Hematol. 2018, 97, 443–451.
- Abdel-Wahab, O.; Manshouri, T.; Patel, J.; Harris, K.; Yao, J.; Hedvat, C.; Heguy, A.; Bueso-Ramos, C.; Kantarjian, H.; Levine, R.L.; et al. Genetic analysis of transforming events that convert chronic myeloproliferative neoplasms to leukemias. Cancer Res. 2010, 70, 447–452.
- Spivak, J.L. How I treat polycythemia vera. Blood 2019, 134, 341–352.
- Antonioli, E.; Guglielmelli, P.; Pieri, L.; Finazzi, M.; Rumi, E.; Martinelli, V.; Vianelli, N.; Luigia Randi, M.; Bertozzi, I.; De Stefano, V.; et al. Hydroxyurea-related toxicity in 3,411 patients with Ph’-negative MPN. Am. J. Hematol. 2012, 87, 552–554.
- Alvarez-Larrán, A.; Kerguelen, A.; Hernández-Boluda, J.C.; Pérez-Encinas, M.; Ferrer-Marín, F.; Bárez, A.; Martínez-López, J.; Cuevas, B.; Mata, M.I.; García-Gutiérrez, V.; et al. Frequency and prognostic value of resistance/intolerance to hydroxycarbamide in 890 patients with polycythaemia vera. Br. J. Haematol. 2016, 172, 786–793.
- Alvarez-Larrán, A.; Díaz-González, A.; Such, E.; Mora, E.; Andrade-Campos, M.; García-Hernández, C.; Gómez-Casares, M.T.; García-Gutiérrez, V.; Carreño-Tarragona, G.; Garrote, M.; et al. Genomic characterization of patients with polycythemia vera developing resistance to hydroxyurea. Leukemia 2021, 35, 623–627.
- Bose, P.; Verstovsek, S. JAK2 inhibitors for myeloproliferative neoplasms: What is next? Blood 2017, 130, 115–125.
- Vannucchi, A.M.; Kiladjian, J.J.; Griesshammer, M.; Masszi, T.; Durrant, S.; Passamonti, F.; Harrison, C.N.; Pane, F.; Zachee, P.; Mesa, R.; et al. Ruxolitinib versus standard therapy for the treatment of polycythemia vera. N. Engl. J. Med. 2015, 372, 426–435.
- Verstovsek, S.; Passamonti, F.; Rambaldi, A.; Barosi, G.; Rosen, P.J.; Rumi, E.; Gattoni, E.; Pieri, L.; Guglielmelli, P.; Elena, C.; et al. A phase 2 study of ruxolitinib, an oral JAK1 and JAK2 inhibitor, in patients with advanced polycythemia vera who are refractory or intolerant to hydroxyurea. Cancer 2014, 120, 513–520.
- Passamonti, F.; Griesshammer, M.; Palandri, F.; Egyed, M.; Benevolo, G.; Devos, T.; Callum, J.; Vannucchi, A.M.; Sivgin, S.; Bensasson, C.; et al. Ruxolitinib for the treatment of inadequately controlled polycythaemia vera without splenomegaly (RESPONSE-2): A randomised, open-label, phase 3b study. Lancet Oncol. 2017, 18, 88–99.
- Verstovsek, S.; Courby, S.; Griesshammer, M.; Mesa, R.A.; Brachmann, C.B.; Kawashima, J.; Maltzman, J.D.; Shao, L.; Xin, Y.; Huang, D.; et al. A phase 2 study of momelotinib, a potent JAK1 and JAK2 inhibitor, in patients with polycythemia vera or essential thrombocythemia. Leuk Res. 2017, 60, 11–17.
- Samuelson, B.T.; Vesely, S.K.; Chai-Adisaksopha, C.; Scott, B.L.; Crowther, M.; Garcia, D. The impact of ruxolitinib on thrombosis in patients with polycythemia vera and myelofibrosis: A meta-analysis. Blood Coagul Fibrinolysis 2016, 27, 648–652.
- Besremi: EPAR—Public Assessment report., European Medicines Agency, December 2018. Available online: (accessed on 25 February 2021).
- Gisslinger, H.; Klade, C.; Georgiev, P.; Krochmalczyk, D.; Gercheva-Kyuchukova, L.; Egyed, M.; Rossiev, V.; Dulicek, P.; Illes, A.; Pylypenko, H.; et al. Ropeginterferon alfa-2b versus standard therapy for polycythaemia vera (PROUD-PV and CONTINUATION-PV): A randomised, non-inferiority, phase 3 trial and its extension study. Lancet Haematol. 2020, 7, e196–e208.
- Barbui, T.; Vannucchi, A.M.; De Stefano, V.; Masciulli, A.; Carobbio, A.; Ferrari, A.; Ghirardi, A.; Rossi, E.; Ciceri, F.; Bonifacio, M.; et al. Ropeginterferon alfa-2b versus phlebotomy in low-risk patients with polycythaemia vera (Low-PV study): A multicentre, randomised phase 2 trial. Lancet Haematol. 2021, 8, e175–e184.
- de Weerd, N.A.; Nguyen, T. The interferons and their receptors--distribution and regulation. Immunol. Cell Biol. 2012, 90, 483–491.
- Chawla-Sarkar, M.; Lindner, D.J.; Liu, Y.-F.; Williams, B.R.; Sen, G.C.; Silverman, R.H.; Borden, E.C. Apoptosis and interferons: Role of interferon-stimulated genes as mediators of apoptosis. Apoptosis 2003, 8, 237–249.
- Hansen, I.O.; Sørensen, A.L.; Hasselbalch, H.C. Second malignancies in hydroxyurea and interferon-treated Philadelphia-negative myeloproliferative neoplasms. Eur. J. Haematol. 2017, 98, 75–84.
- Faille, D.; Lamrani, L.; Loyau, S.; Huisse, M.-G.; Bourrienne, M.-C.; Alkhaier, S.; Cassinat, B.; Boulaftali, Y.; Debus, J.; Jandrot-Perrus, M.; et al. Interferon Alpha Therapy Increases Pro-Thrombotic Biomarkers in Patients with Myeloproliferative Neoplasms. Cancers 2020, 12, 992.
- Iurlo, A.; Cattaneo, D.; Bucelli, C.; Baldini, L. New Perspectives on Polycythemia Vera: From Diagnosis to Therapy. Int. J. Mol. Sci. 2020, 21, 5805.

