 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Myoung Ok Kim | + 4498 word(s) | 4498 | 2021-05-11 11:35:28 | | | |
| 2 | Conner Chen | Meta information modification | 4498 | 2021-05-25 09:23:45 | | | | |
| 3 | Conner Chen | Meta information modification | 4498 | 2021-09-22 03:05:16 | | |
Video Upload Options
T-lymphokine-activated killer cell-originated protein kinase (TOPK, also known as PDZ-binding kinase or PBK) plays a crucial role in cell cycle regulation and mitotic progression. Abnormal overexpression or activation of TOPK has been observed in many cancers, including colorectal cancer, triple-negative breast cancer, and melanoma, and it is associated with increased development, dissemination, and poor clinical outcomes and prognosis in cancer. Moreover, TOPK phosphorylates p38, JNK, ERK, and AKT, which are involved in many cellular functions, and participates in the activation of multiple signaling pathways related to MAPK, PI3K/PTEN/AKT, and NOTCH1; thus, the direct or indirect interactions of TOPK make it a highly attractive yet elusive target for cancer therapy. Small molecule inhibitors targeting TOPK have shown great therapeutic potential in the treatment of cancer both in vitro and in vivo, even in combination with chemotherapy or radiotherapy. Therefore, targeting TOPK could be an important approach for cancer prevention and therapy.
1. TOPK
T-lymphokine-activated killer cell-originated protein kinase (TOPK), also known as PDZ-binding kinase (PBK), is a serine–threonine kinase of the mitogen-activated protein kinase kinase (MAPKK) family and an oncogenic protein that regulates cell survival, proliferation, growth, apoptosis, and inflammation [1][2][3]. TOPK is also a mitosis kinase that is activated by the CDK1/cyclin B1 complex to promote cytokinesis through phosphorylation of polycomb repressive complex 1 (PRC1) [4][5] or anaphase-promoting complex (APC) ubiquitin ligase [6]; inactivation of protein phosphatase 1 alpha (PP1a) leads to phosphorylation at Thr9, which promotes TOPK activation [7]. Activated TOPK leads to chromatin condensation by facilitating phosphorylation of histone H3 at Ser10 in M phase; furthermore, the depletion of TOPK induced by siRNA causes cytokinetic defects in cancer cells [7][8]. Thus, TOPK functions in cancer development. In addition, TOPK has also been recognized as a metastasis-promoting kinase in cancer metastasis [9]. In addition, high expression of TOPK is correlated with oncogenic KRAS and BRAF mutations. Moreover, TOPK could mediate pathway activation independently of B-Raf or C-Raf, which indicates that a TOPK/extracellular signal-regulated kinase 2 (ERK2) feedback loop might bypass the negative feedback loop regulating the Raf/MEK/ERK pathway, which in turn promotes transformation potential. All of those based on ERKs are unique substrates for MEK1 or MEK2, and MEK proteins are downstream targets of Raf kinases [10]. Moreover, TOPK dysregulation can also potentiate cancer development and dissemination. Therefore, given that TOPK is highly transactivated in various types of cancer and related to aggressive tumor phenotypes [11][12][13], it is now considered a promising molecular target for the treatment of malignant tumors.
In terms of its mechanism, overactivation of TOPK promotes cancer cell proliferation, inflammation, and metastasis through activation of downstream signaling cascades, such as the MAPKs and ribosomal S-6 kinase (RSK), as well as transcription factors, including activator protein-1 (AP-1) and NF-κB [14][15][16][17]. Furthermore, TOPK expression is associated with H-Ras-induced cell transformation, activation of c-jun-NH2-kinase (JNK), and p53 expression via UVA, UVB, and DNA damage [18][19], respectively. In addition, TOPK promotes cell migration through regulation of the PI3K/PTEN/AKT-dependent or TGF-beta1/Smad signaling pathways [9][20]. Thus, TOPK performs an oncogenic cellular function, and its inhibition should be effective in cancer therapy. Furthermore, TOPK is a highly ranked radio-sensitizing gene, which further indicates its potential as a target to widen the therapeutic window given its differential expression between cancer and normal tissues [8]. Notably, however, TOPK expression is hardly detectable in normal tissues, except in the testis and fetal tissues. In addition, a TOPK inhibitor is not currently in clinical use. Even the effects of pantoprazole, an FDA-approved inhibitor that targets TOPK, have been reported in a colon cancer only [21]. Subsequent reports of HI-TOPK-032 and OTS964 TOPK inhibitors have indicated disadvantages in terms of solubility and toxicity, which limits the pace of clinical application [22][23]. In summary, TOPK is a novel oncogene that plays a crucial role in the occurrence and development of cancer. Targeting TOPK may help provide new therapeutics for cancer patients; therefore, it is an attractive potential target for the development of chemotherapeutic inhibitors.
2. TOPK Expressed Highly in Cancers and Is a Prognostic Marker of Cancer
The expression of TOPK is significantly higher in various cancers compared with in normal tissues according to The Cancer Genome Atlas database (http://gepia.cancer-pku.cn/) (accessed on 25 March 2021), as shown in Figure 1, with exceptions for head and neck squamous cell carcinoma (HNSC), kidney chromophobe carcinoma (KICH), kidney renal clear cell carcinoma (KIRC), kidney renal clear cell carcinoma (KIRP), pheochromocytoma and paraganglioma (PCPG), prostate adenocarcinoma (PRAD), and thyroid carcinoma (THCA). Notably, TOPK expression is reduced in acute myeloid leukemia (LAML) and testicular germ cell tumors (TGCT) versus normal tissues. Indeed, samples from patients with 24 of 31 (Figure 1A, red pane, * p < 0.05) tumor types had markedly higher expression of TOPK, which further highlights the suitability of TOPK as a target for cancer treatment. By examining the relationship between survival and expression of TOPK (survival data are collected in Figure 1), it becomes clear that high levels of TOPK expression are significantly associated with survival in 11 of 33 tumor samples. Interestingly, bladder urothelial carcinoma (BLCA), breast invasive carcinoma (BRCA), cervical squamous cell carcinoma (CESC), cholangiocarcinoma (CHOL), esophageal carcinoma (ESCA), glioblastoma multiforme (GBM), HNSC, PRAD, stomach adenocarcinoma (STAD), and uterine corpus endometrial carcinoma (UCEC), all of which show TOPK upregulation, are not correlated with survival.
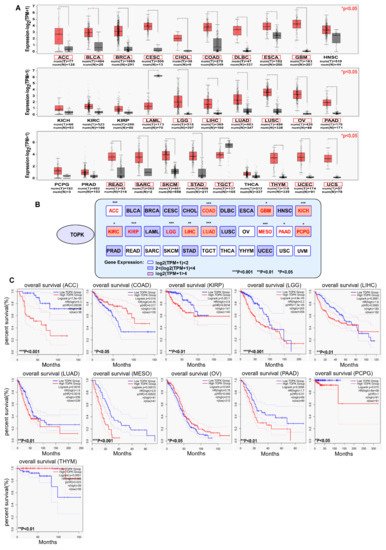
Figure 1. The expression of TOPK in cancers and TOPK expression related to overall survival of patients with cancers. (A) Comparison of the expression of TOPK kinases between tumor and normal tissues. The images and significance are from The Cancer Genome Atlas database. * p < 0.05, ** p < 0.01, and *** p < 0.001. (B) Correlation between TOPK kinase expression and patient overall survival. Red text: gene expression had significant relation with survival; black text: gene expression had no significant relation with survival. The survival data are derived from ULCAN database (http://ualcan.path.uab.edu/) (accessed on 25 March 2021). Samples were categorized into two groups for analysis: high TOPK expression (with TPM values above upper quartile); low/medium TOPK expression (with TPM values below upper quartile). * p < 0.05, ** p < 0.01, *** p < 0.001. (C) The correlation of TOPK expression in 11 types of cancer with clinical outcomes; the other 20 types of cancer showed no significant differences with overall survival (data not shown).
As an oncogene that is overactivated, leading to cancer development at a rapid rate, TOPK overexpression is a common molecular characteristic of human malignancies, and it has been reported in many types of human cancers, including colon, lung, esophageal, prostate, and ovarian cancers [9][11][12][13][24]. High expression of TOPK has also been linked to tumor aggressiveness, invasion, and metastatic spread in various malignancies. Therefore, TOPK expression may be associated with poor prognosis in these cancers under certain circumstances or considered a prognostic marker. Previous reports have shown that a significant relationship exists between TOPK expression and poor prognosis in various cancers, such as prostate, liver, lung, colorectal, and brain cancers [18][22][25][26]. High levels of TOPK expression are significantly associated with poor progression-free survival and overall survival in the early-stage cases of epithelial ovarian cancer [24]. In addition, patients with high TOPK expression levels and non-small cell lung cancer also show poorer overall and recurrence survival [9][27] when compared with patients with low TOPK expression. In clinicopathological patients with oral cancer, TOPK is significantly associated with prognosis in various models [28]. Furthermore, analysis of immunostaining for TOPK expression in cholangiocarcinoma from HCC has revealed that TOPK serves as predictor of survival in cholangiocarcinoma patients [29]. Similarly, immunohistochemical staining in colon cancer patients showed that TOPK expression is associated with clinicopathological features and mainly occurs in the cytoplasm and nucleus [30]. TOPK is also a novel prognostic biomarker and therapeutic target for chordoma; suppression of TOPK leads to significantly reduced chordoma cell proliferation and triggers an increase in apoptosis [31]. Koh et al. reported that TOPK is a biomarker for predicting outcomes in patients with primary central nervous system lymphoma [32]. PBK/TOPK is often overexpressed in gastric cancer; thus, it could be a crucial molecular marker to determine the malignant properties of gastric cancer cells and a target for molecular therapy in gastric cancer patients [33]. Moreover, application of fluorescently labeled TOPK inhibitor is used for cancer-specific imaging in a variety of tumors and can potentially improve patient care [34]; for example, the labeling of a TOPK inhibitor with F-18 has been exploited for positron emission tomography imaging in glioblastoma [35], and TOPK may represent a biomarker for malignant glioma [36]. Additionally, TOPK seems to be a valuable prognostic factor in patients with sporadic CRC with KRAS or BRAF mutations, as well as in patients with metastatic disease who respond to anti-EGFR therapies [37]. Hence, given its cancer-specific expression and known functions, TOPK is a potentially valuable cancer biomarker for patient stratification and risk assessment.
3. TOPK Promotes Proliferation
TOPK is a common cancer molecule in human malignancies, and several studies have revealed that it also plays crucial roles in cellular functions, including cell proliferation, DNA damage repair, the cell cycle, apoptosis, immune responses, and inflammation [7][15][18][30][38][39]. It is clear that TOPK is a potential therapeutic target in cancer. Below, we introduce the application of TOPK as a target in cancer therapy from various perspectives.
The expression of TOPK is closely related to cell malignancy and the malignant potential of tumor cells. Previous reports have indicated that overexpression of TOPK promotes cell proliferation and tumor formation in JB6 epidermal cells both in vitro and in vivo [10][40][41]. In contrast, knockdown of TOPK in cancer cell lines suppresses tumor growth [10][42][43]. Moreover, the overexpression and oncogenic activities of TOPK are reported in cancer types. For example, TOPK expression is enhanced in many tumor cell lines compared with in nontransformed cells; its expression can be induced by IGF-I through the phosphatidylinositol 3-kinase (PI3K)/mTOR pathway and the ERK pathway [44]. It has been suggested that TOPK promotes cell growth and survival and is involved in the AKT and MAPK signaling pathways. Specific to cancers, TOPK is regulated by protein phosphatase 2A (PP2A) and BCR/ABL in leukemia and enhances cell proliferation, indicating that it may be a target of BCR/ABL [45], and has been associated with HTLV-1-transformed T-cell lines and ATLL-derived T-cell lines [17]. TOPK is a modulator of CEBPA activity in FLT3-ITD mutated AML cells; therefore, inhibition of TOPK reduces phosphorylation of CEBPA p42, which then becomes available to bind E2F1 and suppresses the transcriptional activity of E2F1/MYC [46]. Furthermore, suppression of TOPK is effective in inhibiting proliferation and survival in CSC subpopulations of nonsmall-cell lung cancer cells; this is thought to be related to suppression of FOXM1 activity, which plays a crucial role in the expression of late cell cycle control genes [40]. Moreover, given that TOPK interacts with the DNA binding domain (DBD) of tumor suppressor p53 and leads to a decrease in cell cycle regulatory proteins, such as p21, which consequently leads to cancer development and progression [18], PBK/TOPK may be an effective target for antineoplastic kinase inhibitors that induce apoptosis and suppress growth. Therefore, through targeting TOPK could suppress cancer proliferation.
4. TOPK Promotes Tumor Dissemination
TOPK putatively promotes metastasis; plays a role in epithelial–mesenchymal transition; and facilitates tumor invasiveness of lung, prostate, gastric, pancreatic, or breast cancer cells. Previous studies have suggested that TOPK/PBK promotes cell migration through regulation of the PI3K/PTEN/AKT pathway in lung cancer [9]. Jiang et al. (2020) showed that TOPK promotes metastasis via γ-catenin through Src/GSK3β/STAT3 signaling activated in esophageal squamous cell carcinoma. Through upregulation of TBX3 in TGF-β1/Smad signaling, TOPK also facilitates epithelial–mesenchymal transition and invasion of breast cancer cells [20]. In addition, TOPK has fundamental roles in the metastasis of other cancers, including colon cancer and melanoma, which are associated with the AKT, ERK, and β-catenin pathways [13][40]. Knocking down TOPK has been shown to decrease the migration of cancer cells both in vitro and in vivo [9][11][12]. Indeed, TOPK is highly expressed in tumor cells and facilitates metastasis; hence, targeting TOPK could suppress cancer metastasis. In addition, TOPK promotes tumor dissemination by direct phosphorylation of p53-related protein kinase (PRPK) at its Ser250 residue, which in turn regulates the phosphorylation status of p53 and AKT [47]. TOPK expression has also been related to distant metastasis of tumor cells. Studies have reported that TOPK expression in lung cancer is significantly higher than expression in paired paracancerous tissues and benign tumors, which represents an independent risk factor for lymph node metastasis and distant metastasis of lung cancer [43]. Furthermore, the metastatic activity of lung cancer cells (H1299) was increased by 2~3-fold after transfection with TOPK plasmid, whereas knockdown of TOPK inhibited metastasis [9]. Reports are increasingly suggesting that specific drugs can inhibit the activity of TOPK in an ATP-competitive—or non-competitive—manner to reduce cell proliferation and metastasis [2][48]. Thus, TOPK is related to the cancer metastasis.
5. TOPK Regulates the Cell Cycle
TOPK expression and activation levels directly regulate the cell cycle. Indeed, TOPK may be an important hallmark in this progress. TOPK affects the process of cell mitosis and promotes cell division by activating PRC1, which plays an important role in the formation of the spindle and its equatorial plan. Prior studies showed that TOPK could bind to PRC1 through its C-terminal glutamate aspartic acid repetitive sequence to promote the phosphorylation of PRC at the T481 site, which, therefore, increased the phosphorylation level of CDK1/cyclin B1 to PRC1 and eventually promoted cell cycle division [4]. In addition, some studies have reported that TOPK ubiquitinated by CHFR resulted in phosphorylated and inactivated PTEN, consequently inducing the activation of AKT, which is important for cell cycle progression during the G2 to M phase transition [49]. Studies have also revealed that TOPK expression levels and kinase activity are positively correlated with the number of cells at the G2/M phase in gastric carcinoma and prostate cancer, whereas silencing TOPK or inhibiting it activation can arrest the cell cycle in the G0 phase [50][51]. In this process, the cell cycle is arrested via TOPK and the CDK1/cyclin B complex formation, which prolongs the degradation of cyclin B. Meanwhile, the expression time of cyclin B during cell division is prolonged. However, Liu et al. [38] found that knockdown of TOPK resulted in G2/M cell cycle arrest in promyelocytes, a phenomenon that may result in hysteresis during the formation of the CDK1 and cyclin B complex. In another study, the activity of PP1a was inhibited after its phosphorylation (Thr320) by CDK1⁄cyclin B1 in early-to-mid mitosis [52]. Apparently, partial silencing of TOPK was unable to block cell cycle progression but altered the duration of the cell cycle phase. Thus, the TOPK expression pattern on the cell cycle correlated with that of cyclin B1. Furthermore, TOPK is also associated with the S phase in the cell cycle. An in vitro study has shown that TOPK plays a hitherto unknown role during S phase, suggesting that TOPK depletion promotes fork stalling and collapse under conditions of replication stress and exogenous DNA damage, thereby inducing cell apoptosis [53]. Moreover, activated TOPK phosphorylates histone H3 (Ser10) at the M phase, while the H3 histone variant CENP-A is an epigenetic marker critical for centromere identity and function. Yu et al. [54] reported that CDK1 and PP1α control the phosphorylation status of CENP-A Ser68, which orchestrates its cell-cycle-dependent deposition at centromeres. However, the relationship between TOPK and CENP-A at centromeres during the cell cycle remains to be fully explored. Prior studies showed that phosphorylation by PBK/TOPK was impaired when Thr450 was substituted to alanine (T450A) and that this lead to the isolation of homologous chromatids and the formation of F-actin polymerization in contraction rings [55]. Another study indicated that targeting the phosphorylated p97 site of TOPK could lead to mitosis discontinuity [7]. Moreover, TOPK-dependent transcriptional regulation of cyclin B2 was critical for tumorigenesis and radioresistance in GBM cells [56]. These findings suggest that TOPK is a critical target for cancer treatment in the inhibition of mitotic progress.
6. TOPK Induces Apoptosis Resistance in Tumor Cells
The abnormal expression of TOPK is not only associated with the cell cycle but also with apoptosis and its activation; thus, it confers resistance to drug-induced apoptosis and favors carcinogenesis. Previous reports have revealed that TOPK can downregulate the activation of p53 through its DBD. Furthermore, the expression of p53, its target gene, and cyclin-dependent kinase inhibitor p21 are upregulated in TOPK knockdown experiments [18]. Activation of TOPK in cancer may be promoted by inhibiting microRNA-mediated regulation [57]. In addition, restoration of miR-216b-3p expression in cancer cells is sufficient to inhibit proliferation, promote apoptotic cell death, and overcome TOPK-related downregulation of p53 and p21 [58]. Roh et al. [59] reported that PRPK is a novel kinase downstream of TOPK and a critical player in the promotion of skin carcinogenesis; they showed that knockdown of PRPK increased paclitaxel-induced apoptosis. However, the FDA-approved antibiotic cephalosporin [60] and synthetic compound ADA-07 [61] have been shown to suppress skin carcinogenesis by blocking TOPK activity. In addition, knockdown of TOPK inhibits Prx1 (Ser-32) phosphorylation and thereby induces apoptosis that benefits skin cancer therapy [62]; indeed, knockout or mutation of TOPK leads to the phosphorylation of Prx1 and significantly increases H2O2. The ability of cells to tolerate oxidative stress is ultimately decreased, and this triggers apoptosis [63]. Taken together, these findings indicate that targeting TOPK is efficacious for cancer drugs resistance and, therefore, induces cell apoptosis.
7. Others Factors: Inflammation, DNA Damage, and Autophagy
Little is known about the role played by TOPK in inflammation; however, TOPK is reported to be involved in ultraviolet (UV) light-mediated inflammation through phosphorylation of MKP1 and PRPK [3][47]. Furthermore, Seol et al. [15] found that in TOPK-depleted cells, LPS-induced inducible nitric oxide synthase (iNOS) expression was significantly diminished compared to that in control cells, whereas iNOS was induced by activation of TOPK through LPS/TLR4-induced signaling cascades, which suggests that TOPK is involved in LPS-mediated inflammation. This process can, therefore, result in cell migration and invasion in breast cancer [16]. On the other hand, LPS induces immune responses that elicit the production of NO; PGE2; and cytokines, such as TNF-α, IL-1β, and IL-6, in macrophages when its increases TOPK levels and phosphorylates serine or threonine residues in TOPK; this leads to the activation of HDAC1/HDAC2 that contributes to neuroprotective effects against cerebral ischemia reperfusion injury [64]. TOPK activation-mediated anti-inflammation is also suggested to be involved in remote limb ischemic postconditioning invoked protection against renal ischemia/reperfusion injury; thus, TOPK is a promising target for new drug development in the treatment of ischemic stroke.
In addition, PBK knockdown affects the DNA damage response of cells and enhances the sensitivity of cells to genotoxic agents. Previous studies have shown that TOPK acts as mediator of p38 growth-factor activation (which functions in motility), plays a part in the DNA damage sensing machinery, and contributes to γ-H2AX generation, whereas knockdown of TOPK can impair the generation of γ-H2AX [44]. Hence, PBK likely contributes to γ-H2AX generation, which is responsible for the recruitment of DNA damage response proteins to damage sites; however, its function in activation of DNA damage repair machinery may provide tumor cells with a more efficient repair response that would facilitate tumor growth. Furthermore, TOPK binds with and phosphorylates histone H2AX, which suppresses As3+-induced apoptosis in melanoma cells [65], and TOPK silencing reduces the number of γ-H2AX foci in MCF-7 cells following UV-induced DNA damage [66].
TOPK is related to MAPK, PI3K/AKT, mTOR, and other signal transduction pathways involved in autophagy regulation and, therefore, may participate in this process. Prior research has revealed that inhibiting autophagy increases paclitaxel/cisplatin-triggered apoptosis in cancer cells [67][68][69]. Lu et al. [70] reported that TOPK could bind with and phosphorylate ULK1 at the sites Ser469, Ser495, and Ser533, which led to the increased sensitivity of glioma cells to temozolomide by inhibiting autophagy. Ma et al. [71] found that ecotropic viral integration site-1 (EVI1) confers ovarian cancer cells with cisplatin resistance by inducing autophagy through targeting of the TOPK promoter region in ovarian cancer. In addition, microRNA-mediated autophagy regulation in cancer treatment also plays an important role in chemotherapy resistance/sensitivity [72]; for example, a previous report indicated that miR-216b enhanced chemosensitivity by regulating PBK [58]. These studies suggest that TOPK is a novel autophagy regulator and that targeting TOPK benefits the sensitivity of anticancer clinical drugs.
8. TOPK-Interacting Proteins Involved in Signaling Pathways
TOPK is a member of the novel MEK3/6-related MAPKK family that has crucial functions in various cellular processes. Although some studies have reported that ERK1/2 can only be selectively activated by MEK [73][74], Zhu et al. [10] found that ERK could also be activated by TOPK in colon cancer cells. Other studies have now indicated that knockdown of TOPK or treatment with TOPK inhibitors results in decreased ERK activity [1][15][22], while the expressions of c-MYC, NF-κB, and CREB, all of which are downstream of MEK, are also all reduced by such treatments. Furthermore, some typical kinases play crucial roles in many signal transduction pathways, and their activity is tightly regulated by two phosphorylation events [75][76]; thus, it can be seen that the autophosphorylation of proteins is also essential for regulating signaling pathways. Although the structural consequences of the autophosphorylation process of TOPK have not been reported, this role should be noted. TOPK can also activate other families of the MAPK pathway. For example, Dougherty et al. [77] found that TOPK also plays a crucial role in progenitors and is generally associated with the p38/MAPK pathway, indicating that TOPK is an important regulator of growth and self-renewal in progenitor. Furthermore, targeting TOPK could promote apoptosis by inducing ROS and activating the JNK/p38 signaling pathway [78]. Kim et al. [22] used a TOPK inhibitor to study colon cancer and found that phosphorylation of ERK–RSK was reduced by the inhibitor. The novel selective TOPK inhibitor SKLB-C05 has also been reported to block proliferation and metastasis of human CRC by regulating specific TOPK downstream signaling, including ERK1/2, p38, and JNK1/2/3 signaling [48]. Moreover, the activation of EGFR with the treatment of EGF in lung cancer cells was positively correlated with TOPK phosphorylation. Li et al. [79] found that TOPK was significantly upregulated and profoundly activated in lung cancer cells that exhibited resistance to EGFR-TKIs, which phosphorylated c-Jun, leading to an increased level of AP-1 [80]. It is suggested that TOPK is involved in MAPK constitutively activation. In addition, targeting the COX2/MET/TOPK signaling pathway blocks gefitinib resistance through AP-1 in lung cancer [81]. Therefore, TOPK is considered a MAPKK-like protein that is involved in ERK/MAPK, p38MAPK, and JNK signaling, possibly in a cell type-dependent manner.
TOPK is also involved in the PI3K/AKT pathway in tumor development. Recent results have shown that TOPK promotes cell migration by regulating the PI3K/PTEN/AKT pathway, whereas knockdown of TOPK has the reverse effect [9]. Previous studies also indicate that both the Ras-Raf-MEK-ERK and PI3K-AKT signaling pathways are constitutively activated through multiple mechanisms in cancers [14][82], suggesting that cross-talk and compensation exist between the two pathways. In addition, Jiang et al. [12] reported that TOPK promotes ESCC metastasis by activating the Src/GSK3β/STAT3 and ERK signaling pathways. Therefore, TOPK is an important signaling molecule in PI3K/AKT activation of ERK1/2 signaling. Based on these results, we determined the molecules activated and regulated by TOPK. In addition, we summarized TOPK-related proteins, including upstream, downstream, and transcription factors, as previously reported in the literature (Table 1). We then used the STRING database to obtain an interactome network based on TOPK-interacting proteins (i.e., those listed in Table 1; see Figure 2A). Furthermore, we used the Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway and Gene Ontology (GO) enrichment analyses to further investigate these proteins. Our results indicated that TOPK-related proteins are involved in processes including mitosis and cell cycle progression and play important roles in kinase binding. These proteins are either directly or indirectly related to key molecules in crucial signaling pathways, such as the MAPK, FOXO, PI3K–Akt, ad TNF, and p53 pathways (Figure 2B–E).
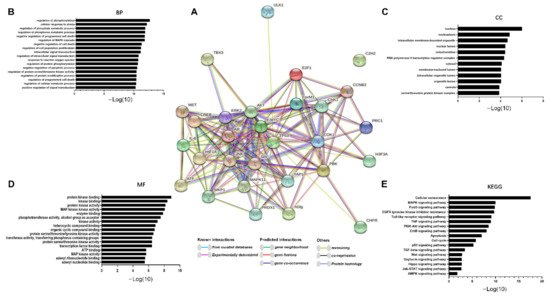
Figure 2. The TOPK interactome and functional enrichment of the network. (A) The interactome in the center was acquired through the STRING database based on the TOPK interaction proteins mentioned in Table 1. (B–D) Functional enrichment of network proteins that interact with TOPK. (E) Signaling pathway enrichment of network proteins that interact with TOPK.
Table 1. Proteins regulated by TOPK involved in various pathways.
| Name | Effect Site | Function | Mechanisms | Ref |
|---|---|---|---|---|
| AKT | Phosphorylation of Ser473 | TOPK promotes AKT phosphorylation at Ser473 and decreases PTEN levels | PTEN–AKT pathway | [9] |
| AR | N/A | promotes proliferation via upregulation of PBK | A reciprocal feedback between TOPK and AR | [83] |
| CDK1 | Binds to #234–275 of TOPK, the C-terminal side of the PRC1-binding sequence | Involved in mitosis | Cell cycle arrest | [4] |
| CHK1 | CHFR ubiquitinates and regulates TOPK | Involved in mitosis | S phase arrest | [53] |
| CHFR | TOPK interacts with CHK1 | Involved in mitosis | G2 to M progression | [49] |
| c-MYC | c-Myc activate TOPK | Represses TOPK transcription | Inhibition of c-Myc- E2F1-PBK signaling that decreases cell growth and survival | [84] |
| CCNB2 | N/A | Represses TOPK transcription | PBK is essential for radioresistance by transcriptional regulation of CCNB2 | [56] |
| C2H2 | Phosphorylate C2H2 linker sequences | Regulate mitosis | Cell cycle arrest | [85] |
| CREB/ATF | Binding the PBK promoter (−312 bp) | Represses TOPK transcription | Associates with TOPK promoter | [86] |
| ERK2 | TOPK–ERK interaction | Increases ERK activity | MAPK–ERK pathway | [10] |
| E2F1 | Binding sites within the PBK promoter (−146 bp) | Represses TOPK transcription | Associates with TOPK promoter | [86] |
| FoxM1 | PBK promoter | Increases the stability and activity of TOPK | FoxM1/PBK/β-catenin signaling | [40] |
| HIF1A | N/A | Transcription of HIF1A increased by TOPK regulation | HIF-1a/snail pathway | [87] |
| H3F3A | Phosphorylation of S10 | Involved in mitosis | Cell cycle arrest | [88] |
| hDlg | Binds to the PDZ2 domain of hDlg | PBK and hDlg are phosphorylated at mitosis; phosphorylation of PBK is required for its kinase activity | PBK could link hDlg or other PDZ-containing proteins to signal transduction pathways regulating the cell cycle or cellular proliferation | [88] |
| IL-6 | N/A | Increases TOPK activity | Cell cycle arrest | [89] |
| JNK | Binds to the JNK | TOPK regulates UVB-induced JNK1 activity | TOPK enhanced the ability of JNKs to mediate H-Ras-induced cell transformation | [19] |
| LGN/GPSM2 | Thr450 | Mitosis activity | Cell cycle arrest | [55] |
| MAPK11 | TOPK-P38 interaction | Mitosis activity | p38MAPK activity and regulation of the DNA damage response | [44] |
| MET | Phosphorylates Y74 of TOPK | Increases the activity of TOPK | COX2/MET/TOPK signaling pathway | [81] |
| MKP1 | Phosphorylation of Ser-359 | TOPK directly binds with and phosphorylates MKP1 | p38 signaling pathway |
[3] |
| NF-κB | N/A | Represses TOPK transcription | ERK pathway | [22] |
| NF-κB | N/A | Activated by TOPK | Upregulation of snail/slug in TGF-β1 signaling | [90] |
| PRDX1 | Ser32 | TOPK interacts with and phosphorylates Prx1 | Induced apoptosis | [62] |
| PRC1 | T481 | TOPK interacts with and phosphorylates PRC1 | Stopping formation of mitotic spindles and spindle midzone | [4] |
| RORγ | N/A | PBK interacts with RORγ and modulates RORγ protein stability | AR-PBK- RORγ-AR pathway | [91] |
| SRC | Phosphorylation of TOPK at Y74 and Y272 | Increases the stability and activity of TOPK | TOPK–histone H3 pathway | [42] |
| TP53 | TOPK interacts with p53 through a DNA-binding domain | PBK modulation of p21 expression is dependent on p53 | Cell cycle arrest | [18] |
| TBX3 | TBX3 was activated by TOPK | TOPK upregulates T-box transcription factor TBX3 | TGF-b1/Smad signaling | [20] |
| ULK1 | Ser469, Ser495, and Ser533 | TOPK binds and phosphorylates ULK1 | Promoting ubiquitination degradation of ULK1 | [70] |
| YAP1 | N/A | Represses TOPK transcription | Hippo-YAP/TAZ pathway | [92] |
| γ-catenin | N/A | TOPK binds with γ-catenin | Src/GSK3β/STAT3 pathway | [12] |
References
- Zhao, R.; Choi, B.Y.; Wei, L.X.; Fredimoses, M.; Yin, F.X.; Fu, X.R.; Chen, H.Y.; Liu, K.D.; Kundu, J.K.; Dong, Z.G.; et al. Acetylshikonin sup-pressed growth of colorectal tumour tissue and cells by inhibiting the intracellular kinase, T-lymphokine-activated killer cell-originated protein kinase. Br. J. Pharmacol. 2020, 177, 2303–2319.
- Zhao, R.; Huang, H.; Choi, B.Y.; Liu, X.; Zhang, M.; Zhou, S.; Song, M.; Yin, F.; Chen, H.; Shim, J.-H.; et al. Cell growth inhibition by 3-deoxysappanchalcone is mediated by directly targeting the TOPK signaling pathway in colon cancer. Phytomedicine 2019, 61, 152813.
- Li, S.Q.; Zhu, F.; Zykova, T.; Kim, M.O.; Cho, Y.Y.; Bode, A.M.; Peng, C.; Ma, W.Y.; Carper, A.; Langfald, A.; et al. T-LAK Cell-originated Protein Kinase (TOPK) Phosphorylation of MKP1 Protein Prevents Solar Ultraviolet Light-induced Inflammation through In-hibition of the p38 Protein Signaling Pathway. J. Biol. Chem. 2011, 286, 29601–29609.
- Abe, Y.; Takeuchi, T.; Kagawa-Miki, L.; Ueda, N.; Shigemoto, K.; Yasukawa, M.; Kito, K. A Mitotic Kinase TOPK Enhances Cdk1/cyclin B1-dependent Phosphorylation of PRC1 and Promotes Cytokinesis. J. Mol. Biol. 2007, 370, 231–245.
- Matsumoto, S.; Abe, Y.; Fujibuchi, T.; Takeuchi, T.; Kito, K.; Ueda, N.; Shigemoto, K.; Gyo, K. Characterization of a MAPKK-like protein kinase TOPK. Biochem. Biophys. Res. Commun. 2004, 325, 997–1004.
- Kraft, C.; Herzog, F.; Gieffers, C.; Mechtler, K.; Hagting, A.; Pines, J.; Peters, J. Mitotic regulation of the human anaphase-promoting complex by phosphorylation. EMBO J. 2003, 22, 6598–6609.
- Park, J.-H.; Nishidate, T.; Nakamura, Y.; Katagiri, T. Critical roles of T-LAK cell-originated protein kinase in cytokinesis. Cancer Sci. 2010, 101, 403–411.
- Pirovano, G.; Ashton, T.M.; Herbert, K.J.; Bryant, R.J.; Verrill, C.L.; Cerundolo, L.; Buffa, F.M.; Prevo, R.; Harrap, I.; Ryan, A.J.; et al. TOPK modulates tumour-specific radiosensitivity and correlates with recurrence after prostate radiotherapy. Br. J. Cancer 2017, 117, 503–512.
- Shih, M.C.; Chen, J.Y.; Wu, Y.C.; Jan, Y.H.; Yang, B.M.; Lu, P.J.; Cheng, H.C.; Huang, M.S.; Yang, C.J.; Hsiao, M.; et al. TOPK/PBK promotes cell migration via modulation of the PI3K/PTEN/AKT pathway and is associated with poor prognosis in lung cancer. Oncogene 2012, 31, 2389–2400.
- Zhu, F.; Zykova, T.A.; Kang, B.S.; Wang, Z.; Ebeling, M.C.; Abe, Y.; Ma, W.; Bode, A.M.; Dong, Z. Bidirectional Signals Transduced by TOPK-ERK Interaction Increase Tumorigenesis of HCT116 Colorectal Cancer Cells. Gastroenterology 2007, 133, 219–231.
- Zykova, T.A.; Zhu, F.; Wang, L.; Li, H.; Bai, R.; Lim, Y.; Yao, K.; Bode, A.M.; Dong, Z. The T-LAK Cell-originated Protein Kinase Signal Pathway Promotes Colorectal Cancer Metastasis. EBioMedicine 2017, 18, 73–82.
- Jiang, Y.A.; Zhang, J.; Zhao, J.M.; Li, Z.Z.; Chen, H.Y.; Qiao, Y.; Chen, X.H.; Liu, K.D.; Dong, Z.M. TOPK promotes metastasis of esophageal squamous cell carcinoma by activating the Src/GSK3 beta/STAT3 signaling pathway via gamma-catenin. BMC Cancer 2020, 20, 1145.
- Sun, H.; Zhang, L.; Shi, C.; Hu, P.; Yan, W.; Wang, Z.; Duan, Q.; Lu, F.; Qin, L.; Lu, T.; et al. TOPK is highly expressed in circulating tumor cells, enabling metastasis of prostate cancer. Oncotarget 2015, 6, 12392–12404.
- Aksamitiene, E.; Kholodenko, B.N.; Kolch, W.; Hoek, J.B.; Kiyatkin, A. PI3K/Akt-sensitive MEK-independent compensatory circuit of ERK activation in ER-positive PI3K-mutant T47D breast cancer cells. Cell. Signal. 2010, 22, 1369–1378.
- Park, J.H.; Jeong, Y.J.; Won, H.K.; Choi, S.Y.; Park, J.H.; Oh, S.M. Activation of TOPK by lipopolysaccharide promotes induction of inducible nitric oxide synthase through NF-kappaB activity in leukemia cells. Cell. Signal. 2014, 26, 849–856.
- Seol, M.A.; Park, J.H.; Jeong, J.H.; Lyu, J.; Han, S.Y.; Oh, S.M. Role of TOPK in lipopolysaccharide-induced breast cancer cell mi-gration and invasion. Oncotarget 2017, 8, 40190–40203.
- Ishikawa, C.; Senba, M.; Mori, N. Mitotic kinase PBK/TOPK as a therapeutic target for adult T-cell leukemia/lymphoma. Int. J. Oncol. 2018, 53, 801–814.
- Hu, F.; Gartenhaus, R.B.; Eichberg, D.; Liu, Z.; Fang, H.B. Rapoport AP: PBK/TOPK interacts with the DBD domain of tumor sup-pressor p53 and modulates expression of transcriptional targets including p21. Oncogene 2010, 29, 5464–5474.
- Oh, S.-M.; Zhu, F.; Cho, Y.-Y.; Lee, K.W.; Kang, B.S.; Kim, H.-G.; Zykova, T.; Bode, A.M.; Dong, Z. T-Lymphokine–Activated Killer Cell–Originated Protein Kinase Functions as a Positive Regulator of c-Jun-NH2-Kinase 1 Signaling and H-Ras–Induced Cell Transformation. Cancer Res. 2007, 67, 5186–5194.
- Lee, Y.J.; Park, J.H.; Oh, S.M. TOPK promotes epithelial-mesenchymal transition and invasion of breast cancer cells through up-regulation of TBX3 in TGF-beta 1/Smad signaling. Biochem. Biophys. Res. Commun. 2020, 522, 270–277.
- Zeng, X.; Liu, L.; Zheng, M.; Sun, H.; Xiao, J.; Lu, T.; Huang, G.; Chen, P.; Zhang, J.; Zhu, F.; et al. Pantoprazole, an FDA-approved proton-pump inhibitor, suppresses colorectal cancer growth by targeting T-cell-originated protein kinase. Oncotarget 2016, 7, 22460–22473.
- Kim, D.J.; Li, Y.; Reddy, K.; Lee, M.-H.; Kim, M.O.; Cho, Y.-Y.; Lee, S.-Y.; Kim, J.-E.; Bode, A.M.; Dong, Z. Novel TOPK Inhibitor HI-TOPK-032 Effectively Suppresses Colon Cancer Growth. Cancer Res. 2012, 72, 3060–3068.
- Matsuo, Y.; Park, J.-H.; Miyamoto, T.; Yamamoto, S.; Hisada, S.; Alachkar, H.; Nakamura, Y. TOPK inhibitor induces complete tumor regression in xenograft models of human cancer through inhibition of cytokinesis. Sci. Transl. Med. 2014, 6, 259ra145.
- Ikeda, Y.; Park, J.-H.; Miyamoto, T.; Takamatsu, N.; Kato, T.; Iwasa, A.; Okabe, S.; Imai, Y.; Fujiwara, K.; Nakamura, Y.; et al. T-LAK Cell-Originated Protein Kinase (TOPK) as a Prognostic Factor and a Potential Therapeutic Target in Ovarian Cancer. Clin. Cancer Res. 2016, 22, 6110–6117.
- Park, J.H.; Lin, M.L.; Nishidate, T.; Nakamura, Y.; Katagiri, T. PDZ-binding kinase/T-LAK cell-originated protein kinase, a putative cancer/testis antigen with an oncogenic activity in breast cancer. Cancer Res. 2006, 66, 9186–9195.
- Herrero-Martín, D.; Osuna, D.; Ordóñez, J.L.; Sevillano, V.; Martins, A.S.; Mackintosh, C.; Campos, M.; Madoz-Gúrpide, J.; Otero-Motta, A.P.; Caballero, G.; et al. Stable interference of EWS–FLI1 in an Ewing sarcoma cell line impairs IGF-1/IGF-1R signalling and reveals TOPK as a new target. Br. J. Cancer 2009, 101, 80–90.
- Lei, B.; Liu, S.; Qi, W.; Zhao, Y.; Li, Y.; Lin, N.; Xu, X.; Zhi, C.; Mei, J.; Yan, Z.; et al. PBK/TOPK expression in non-small-cell lung cancer: Its correlation and prognostic significance with Ki67 and p53 expression. Histopathology 2013, 63, 696–703.
- Chang, C.-F.; Chen, S.-L.; Sung, W.-W.; Hsieh, M.-J.; Hsu, H.-T.; Chen, L.-H.; Chen, M.-K.; Ko, J.-L.; Chen, C.-J.; Chou, M.-C. PBK/TOPK Expression Predicts Prognosis in Oral Cancer. Int. J. Mol. Sci. 2016, 17, 1007.
- He, F.R.; Yan, Q.G.; Fan, L.N.; Liu, Y.X.; Cui, J.H.; Wang, J.H.; Wang, L.; Wang, Y.M.; Wang, Z.; Guo, Y.; et al. PBK/TOPK in the differential diagnosis of cholangiocarcinoma from hepatocellular carcinoma and its involvement in prognosis of human cholangiocarci-noma. Hum. Pathol. 2010, 41, 415–424.
- Su, T.-C.; Chen, C.-Y.; Tsai, W.-C.; Hsu, H.-T.; Yen, H.-H.; Sung, W.-W.; Chen, C.-J. Cytoplasmic, nuclear, and total PBK/TOPK expression is associated with prognosis in colorectal cancer patients: A retrospective analysis based on immunohistochemistry stain of tissue microarrays. PLoS ONE 2018, 13, e0204866.
- Thanindratarn, P.; Dean, D.C.; Nelson, S.D.; Hornicek, F.J.; Duan, Z.F. T-LAK cell-originated protein kinase (TOPK) is a Novel Prognostic and Therapeutic Target in Chordoma. Cell Prolif. 2020, 53, e12901.
- Koh, M.; Hayakawa, Y.; Akai, T.; Hayashi, T.; Tomita, T.; Nagai, S.; Kuroda, S. Novel biomarker, phosphorylated T-LAK cell-originated protein kinase (p-TOPK) can predict outcome in primary central nervous system lymphoma. Neuropathology 2018, 38, 228–236.
- Ohashi, T.; Komatsu, S.; Ichikawa, D.; Miyamae, M.; Okajima, W.; Imamura, T.; Kiuchi, J.; Kosuga, T.; Konishi, H.; Shiozaki, A.; et al. Overexpression of PBK/TOPK relates to tumour malignant potential and poor outcome of gastric carcinoma. Br. J. Cancer 2016, 116, 218–226.
- Pirovano, G.; Roberts, S.; Reiner, T. TOPKi-NBD: A fluorescent small molecule for tumor imaging. Eur. J. Nucl. Med. Mol. Imaging 2020, 47, 1003–1010.
- Pirovano, G.; Roberts, S.; Brand, C.; Donabedian, P.L.; Mason, C.; De Souza, P.D.; Higgins, G.S.; Reiner, T. [18F]FE-OTS964: A Small Molecule Targeting TOPK for In Vivo PET Imaging in a Glioblastoma Xenograft Model. Mol. Imaging Biol. 2018, 21, 705–712.
- Hayashi, T.; Hayakawa, Y.; Koh, M.; Tomita, T.; Nagai, S.; Kashiwazaki, D.; Sugimori, M.; Origasa, H.; Kuroda, S. Impact of a novel biomarker, T-LAK cell-originating protein kinase (TOPK) expression on outcome in malignant glioma. Neuropathology 2018, 38, 144–153.
- Zlobec, I.; Molinari, F.; Kovac, M.; Bihl, M.P.; Altermatt, H.J.; Diebold, J.; Frick, H.; Germer, M.; Horcic, M.; Montani, M.; et al. Prognostic and predictive value of TOPK stratified by KRAS and BRAF gene alterations in sporadic, hereditary and metastatic colorectal cancer patients. Br. J. Cancer 2009, 102, 151–161.
- Liu, Y.; Liu, H.; Cao, H.; Song, B.; Zhang, W.; Zhang, W. PBK/TOPK mediates promyelocyte proliferation via Nrf2-regulated cell cycle progression and apoptosis. Oncol. Rep. 2015, 34, 3288–3296.
- Gao, S.M.; Zhu, Y.; Li, H.B.; Xia, Z.Y.; Wu, Q.P.; Yao, S.L.; Wang, T.T.; Yuan, S.Y. Remote ischemic postconditioning protects against renal ischemia/reperfusion injury by activation of T-LAK-cell-originated protein kinase (TOPK)/PTEN/Akt signaling pathway me-diated anti-oxidation and anti-inflammation. Int. Immunopharmacol. 2016, 38, 395–401.
- Yang, Y.F.; Pan, Y.H.; Cao, Y.; Fu, J.; Yang, X.; Zhang, M.F.; Tian, Q.H. PDZ binding kinase, regulated by FoxM1, enhances malignant phenotype via activation of beta-Catenin signaling in hepatocellular carcinoma. Oncotarget 2017, 8, 47195–47205.
- Fan, X.; Tao, J.; Cai, X.; Fredimoses, M.; Wu, J.; Jiang, Z.; Zhang, X.; Li, S. Eupafolin Suppresses Esophagus Cancer Growth by Targeting T-LAK Cell-Originated Protein Kinase. Front. Pharmacol. 2019, 10, 1248.
- Xiao, J.J.; Duan, Q.H.; Wang, Z.; Yan, W.; Sun, H.M.; Xue, P.P.; Fan, X.M.; Zeng, X.Y.; Chen, J.; Shao, C.; et al. Phosphorylation of TOPK at Y74, Y272 by Src increases the stability of TOPK and promotes tumorigenesis of colon cancer. Oncotarget 2016, 7, 24483–24494.
- Wei, D.-C.; Yeh, Y.-C.; Hung, J.-J.; Chou, T.-Y.; Wu, Y.-C.; Lu, P.-J.; Cheng, H.-C.; Hsu, Y.-L.; Kuo, Y.-L.; Chen, K.-Y.; et al. Overexpression of T-LAK cell-originated protein kinase predicts poor prognosis in patients with stage I lung adenocarcinoma. Cancer Sci. 2012, 103, 731–738.
- Ayllon, V.; Oconnor, R. PBK/TOPK promotes tumour cell proliferation through p38 MAPK activity and regulation of the DNA damage response. Oncogene 2006, 26, 3451–3461.
- Uchida, E.; Suwa, S.; Yoshimoto, R.; Watanabe, K.; Kasama, T.; Miura, O.; Fukuda, T. TOPK is regulated by PP2A and BCR/ABL in leukemia and enhances cell proliferation. Int. J. Oncol. 2019, 54, 1785–1796.
- Alachkar, H.; Mutonga, M.; Malnassy, G.; Park, J.-H.; Fulton, N.; Woods, A.; Meng, L.; Kline, J.; Raca, G.; Odenike, O.; et al. T-LAK cell-originated protein kinase presents a novel therapeutic target inFLT3-ITD mutated acute myeloid leukemia. Oncotarget 2015, 6, 33410–33425.
- Roh, E.; Lee, M.-H.; Zykova, T.A.; Zhu, F.; Nadas, J.; Kim, H.-G.; Bae, K.B.; Li, Y.; Cho, Y.Y.; Curiel-Lewandrowski, C.; et al. Targeting PRPK and TOPK for skin cancer prevention and therapy. Oncogene 2018, 37, 5633–5647.
- Gao, T.; Hu, Q.; Hu, X.; Lei, Q.; Feng, Z.; Yu, X.; Peng, C.; Song, X.; He, H.; Xu, Y.; et al. Novel selective TOPK inhibitor SKLB-C05 inhibits colorectal carcinoma growth and metastasis. Cancer Lett. 2019, 445, 11–23.
- Shinde, S.R.; Gangula, N.R.; Kavela, S.; Pandey, V.; Maddika, S. TOPK and PTEN participate in CHFR mediated mitotic check-point. Cell Sign. 2013, 25, 2511–2517.
- Ohashi, T.; Komatsu, S.; Ichikawa, D.; Miyamae, M.; Okajima, W.; Imamura, T.; Kiuchi, J.; Nishibeppu, K.; Kosuga, T.; Konishi, H.; et al. Overexpression of PBK/TOPK Contributes to Tumor Development and Poor Outcome of Esophageal Squamous Cell Carcinoma. Anticancer. Res. 2016, 36, 6457–6466.
- Li, M.; Chen, J.; Li, X.; Luo, Y.; Fang, Y. The Regulation Role of PBK in Cell Cycle and Apoptosis of Prostate Cancer. Biomed. J. Sci. Tech. Res. 2019, 15, 12375–12379.
- Kwon, Y.G.; Lee, S.Y.; Choi, Y.W.; Greengard, P.; Nairn, A.C. Cell cycle-dependent phosphorylation of mammalian protein phos-phatase 1 by cdc2 kinase. Proc. Natl. Acad. Sci. USA 1997, 94, 2168–2173.
- Herbert, K.J.; Puliyadi, R.; Prevo, R.; Rodriguez-Berriguete, G.; Ryan, A.; Ramadan, K.; Higgins, G.S. Targeting TOPK sensitises tumour cells to radiation-induced damage by enhancing replication stress. Cell Death Differ. 2020, 28, 1333–1346.
- Yu, Z.; Zhou, X.; Wang, W.; Deng, W.; Fang, J.; Hu, H.; Wang, Z.; Li, S.; Cui, L.; Shen, J.; et al. Dynamic Phosphorylation of CENP-A at Ser68 Orchestrates Its Cell-Cycle-Dependent Deposition at Centromeres. Dev. Cell 2015, 32, 68–81.
- Fukukawa, C.; Ueda, K.; Nishidate, T.; Katagiri, T.; Nakamura, Y. Critical roles of LGN/GPSM2 phosphorylation by PBK/TOPK in cell division of breast cancer cells. Genes Chromosom. Cancer 2010, 49, 861–872.
- Mao, P.; Bao, G.; Wang, Y.-C.; Du, C.-W.; Yu, X.; Guo, X.-Y.; Li, R.-C.; Wang, M.-D. PDZ-Binding Kinase-Dependent Transcriptional Regulation of CCNB2 Promotes Tumorigenesis and Radio-Resistance in Glioblastoma. Transl. Oncol. 2020, 13, 287–294.
- Chai, Y.Q.; Xue, H.J.; Wu, Y.M.; Du, X.M.; Zhang, Z.H.; Zhang, Y.L.; Zhang, L.L.; Zhang, S.B.; Zhang, Z.G.; Xue, Z.W. MicroRNA-216b-3p inhibits lung adenocarcinoma cell growth via regulating PDZ binding kinase/T-LAK-cell-originated protein kinase. Exp. Ther. Med. 2018, 15, 4822–4828.
- Zou, J.; Kuang, W.H.; Hu, J.L.; Rao, H.M. miR-216b promotes cell growth and enhances chemosensitivity of colorectal cancer by suppressing PDZ-binding kinase. Biochem. Biophys. Res. Commun. 2017, 488, 247–252.
- Peterson, D.; Lee, J.; Lei, X.C.; Forrest, W.F.; Davis, D.P.; Jackson, P.K.; Belmont, L.D. A Chemosensitization Screen Identifies TP53RK, a Kinase that Restrains Apoptosis after Mitotic Stress. Cancer Res. 2010, 70, 6325–6335.
- Fan, X.; Duan, Q.; Ke, C.; Zhang, G.; Xiao, J.; Wu, D.; Zeng, X.; Chen, J.; Guo, J.; Zhou, J.; et al. Cefradine blocks solar-ultraviolet induced skin inflammation through direct inhibition of T-LAK cell-originated protein kinase. Oncotarget 2016, 7, 24633–24645.
- Gao, G.; Zhang, T.; Wang, Q.; Reddy, K.; Chen, H.; Yao, K.; Wang, K.; Roh, E.; Zykova, T.; Ma, W.; et al. ADA-07 Suppresses Solar Ul-traviolet-Induced Skin Carcinogenesis by Directly Inhibiting TOPK. Mol. Cancer Ther. 2017, 16, 1843–1854.
- Zykova, T.A.; Zhu, F.; Vakorina, T.I.; Zhang, J.; Higgins, L.A.; Urusova, D.V.; Bode, A.M.; Dong, Z. T-LAK Cell-originated Protein Kinase (TOPK) Phosphorylation of Prx1 at Ser-32 Prevents UVB-induced Apoptosis in RPMI7951 Melanoma Cells through the Regulation of Prx1 Peroxidase Activity. J. Biol. Chem. 2010, 285, 29138–29146.
- Calabrese, G.; Peker, E.; Amponsah, P.S.; Hoehne, M.N.; Riemer, T.; Mai, M.; Bienert, G.P.; Deponte, M.; Morgan, B.; Riemer, J. Hyperox-idation of mitochondrial peroxiredoxin limits H2O2 -induced cell death in yeast. EMBO J. 2019, 38, e101552.
- Stadler, J.; Harbrecht, B.G.; Di Silvio, M.; Curran, R.D.; Jordan, M.L.; Simmons, R.L.; Billiar, T.R. Endogenous nitric oxide inhibits the synthesis of cyclooxygenase products and interleukin-6 by rat Kupffer cells. J. Leukoc. Biol. 1993, 53, 165–172.
- Zykova, T.A.; Zhu, F.; Lu, C.; Higgins, L.; Tatsumi, Y.; Abe, Y.; Bode, A.M.; Dong, Z. Lymphokine-Activated Killer T-Cell-Originated Protein Kinase Phosphorylation of Histone H2AX Prevents Arsenite-Induced Apoptosis in RPMI7951 Melanoma Cells. Clin. Cancer Res. 2006, 12, 6884–6893.
- Nandi, A.K.; Ford, T.; Fleksher, D.; Neuman, B.; Rapoport, A.P. Attenuation of DNA damage checkpoint by PBK, a novel mitotic kinase, involves protein–protein interaction with tumor suppressor p53. Biochem. Biophys. Res. Commun. 2007, 358, 181–188.
- Xi, G.; Hu, X.; Wu, B.; Jiang, H.; Young, C.Y.; Pang, Y.; Yuan, H. Autophagy inhibition promotes paclitaxel-induced apoptosis in cancer cells. Cancer Lett. 2011, 307, 141–148.
- Chan, K.K.; Wong, O.G.W.; Wong, E.S.Y.; Chan, K.K.L.; Ip, P.P.C.; Tse, K.Y.; Cheung, A.N.Y. Impact of iASPP on chemoresistance through PLK1 and autophagy in ovarian clear cell carcinoma. Int. J. Cancer 2018, 143, 1456–1469.
- Zamame Ramirez, J.A.; Romagnoli, G.G.; Kaneno, R. Inhibiting autophagy to prevent drug resistance and improve anti-tumor therapy. Life Sci. 2021, 265, 118745.
- Lu, H.; Xiao, J.J.; Ke, C.S.; Ni, X.F.; Xiu, R.J.; Tian, Q.; Pan, H.X.; Zou, L.; Wang, F.; Ma, T.F.; et al. TOPK inhibits autophagy by phosphor-ylating ULK1 and promotes glioma resistance to TMZ. Cell Death Dis. 2019, 10, 1354.
- Ma, H.L.; Li, Y.W.; Wang, X.X.; Wu, H.; Qi, G.H.; Li, R.R.; Yang, N.; Gao, M.; Yan, S.; Yuan, C.Z.; et al. PBK, targeted by EVI1, promotes metastasis and confers cisplatin resistance through inducing autophagy in high-grade serous ovarian carcinoma. Cell Death Dis. 2019, 10, 1714.
- Ashrafizadeh, M.; Zarrabi, A.; Orouei, S.; Kiavash, H.; Hakimi, A.; Amirhossein, Z.; Daneshi, S.; Samarghandian, S.; Baradaran, B.; Najafi, M. MicroRNA-mediated autophagy regulation in cancer therapy: The role in chemoresistance/chemosensitivity. Eur. J. Pharmacol. 2021, 892, 173660.
- Bluhm, B.; Ehlen, H.W.A.; Holzer, T.; Georgieva, V.S.; Heilig, J.; Pitzler, L.; Etich, J.; Bortecen, T.; Frie, C.; Probst, K.; et al. miR-322 stabilizes MEK1 expression to inhibit RAF/MEK/ERK pathway activation in cartilage. Development 2017, 144, 3562–3577.
- Hong, S.K.; Wu, P.K.; Park, J.I. A cellular threshold for active ERK1/2 levels determines Raf/MEK/ERK-mediated growth arrest versus death responses. Cell Sign. 2018, 42, 11–20.
- Thirukkumaran, O.M.; Kluba, M.; Hofkens, J.; Mizuno, H. Autophosphorylation of EGFR at Y954 Facilitated Homodimerization and Enhanced Downstream Signals. Biophys. J. 2020, 119, 2127–2137.
- Boczek, E.E.; Luo, Q.; Dehling, M.; Röpke, M.; Mader, S.L.; Seidl, A.; Kaila, V.R.I.; Buchner, J. Autophosphorylation activates c-Src kinase through global structural rearrangements. J. Biol. Chem. 2019, 294, 13186–13197.
- Dougherty, J.D.; Garcia, A.D.R.; Nakano, I.; Livingstone, M.; Norris, B.; Polakiewicz, R.; Wexler, E.M.; Sofroniew, M.V.; Kornblum, H.I.; Geschwind, D.H. PBK/TOPK, a Proliferating Neural Progenitor-Specific Mitogen-Activated Protein Kinase Kinase. J. Neurosci. 2005, 25, 10773–10785.
- Wang, M.Y.; Lin, Z.R.; Cao, Y.; Zheng, L.S.; Peng, L.X.; Sun, R.; Meng, D.F.; Xie, P.; Yang, J.P.; Cao, L.; et al. PDZ binding kinase (PBK) is a theranostic target for nasopharyngeal carcinoma: Driving tumor growth via ROS signaling and correlating with patient sur-vival. Oncotarget 2016, 7, 26604–26616.
- Li, Y.; Yang, Z.; Li, W.; Xu, S.; Wang, T.; Wang, T.; Niu, M.; Zhang, S.; Jia, L.; Li, S. TOPK promotes lung cancer resistance to EGFR tyrosine kinase inhibitors by phosphorylating and activating c-Jun. Oncotarget 2016, 7, 6748–6764.
- Mansour, S.; Matten, W.; Hermann, A.; Candia, J.; Rong, S.; Fukasawa, K.; Woude, G.V.; Ahn, N. Transformation of mammalian cells by constitutively active MAP kinase kinase. Science 1994, 265, 966–970.
- Xiao, J.; Wang, F.; Lu, H.; Xu, S.; Zou, L.; Tian, Q.; Fu, Y.; Lin, X.; Liu, L.; Yuan, P.; et al. Targeting the COX2/MET/TOPK signaling axis induces apoptosis in gefitinib-resistant NSCLC cells. Cell Death Dis. 2019, 10, 1343.
- Meier, F.; Schittek, B.; Busch, S.; Garbe, C.; Smalley, K.; Satyamoorthy, K.; Li, G.; Herlyn, M. The Ras/Raf/MEK/ERK and PI3K/AKT signaling pathways present molecular targets for the effective treatment of advanced melanoma. Front. Biosci. Landmark 2005, 10, 2986–3001.
- Warren, A.Y.; Massie, C.E.; Watt, K.; Luko, K.; Orafidiya, F.; Selth, L.A.; Mohammed, H.; Chohan, B.S.; Menon, S.; Baridi, A.; et al. A re-ciprocal feedback between the PDZ binding kinase and androgen receptor drives prostate cancer. Oncogene 2019, 38, 1136–1150.
- Hu, F.; Gartenhaus, R.B.; Zhao, X.F.; Fang, H.B.; Minkove, S.; Poss, D.E.; Rapoport, A.P. c-Myc and E2F1 drive PBK/TOPK expression in high-grade malignant lymphomas. Leuk. Res. 2013, 37, 447–454.
- Rizkallah, R.; Batsomboon, P.; Dudley, G.B.; Hurt, M.M. Identification of the oncogenic kinase TOPK/PBK as a master mitotic reg-ulator of C2H2 zinc finger proteins. Oncotarget 2015, 6, 1446–1461.
- Nandi, A.K.; Rapoport, A.P. Expression of PDZ-binding kinase (PBK) is regulated by cell cycle-specific transcription factors E2F and CREB/ATF. Leuk. Res. 2006, 30, 437–447.
- Park, J.H.; Moon, M.; Kim, J.S.; Oh, S.M. TOPK mediates hypoxia-induced epithelial-mesenchymal transition and the invasion of nonsmall-cell lung cancer cells via the HIF-1alpha/snail axis. Biochem. Biophys. Res. Commun. 2021, 534, 941–949.
- Gaudet, S.; Branton, D.; Lue, R.A. Characterization of PDZ-binding kinase, a mitotic kinase. Proc. Natl. Acad. Sci. USA 2000, 97, 5167–5172.
- Côté, S.; Simard, C.; Lemieux, R. Regulation of growth-related genes by interleukin-6 in murine myeloma cells. Cytokine 2002, 20, 113–120.
- Lee, Y.J.; Park, J.H.; Oh, S.M. Activation of NF-kappaB by TOPK upregulates Snail/Slug expression in TGF-beta1 signaling to induce epithelial-mesenchymal transition and invasion of breast cancer cells. Biochem. Biophys. Res. Commun. 2020, 530, 122–129.
- Zhang, X.; Huang, Z.; Wang, J.; Ma, Z.; Yang, J.; Corey, E.; Evans, C.; Yu, A.-M.; Chen, H.-W. Targeting Feedforward Loops Formed by Nuclear Receptor RORγ and Kinase PBK in mCRPC with Hyperactive AR Signaling. Cancers 2021, 13, 1672.
- Dou, X.; Wei, J.; Sun, A.; Shao, G.; Childress, C.; Yang, W.; Lin, Q. PBK/TOPK mediates geranylgeranylation signaling for breast cancer cell proliferation. Cancer Cell Int. 2015, 15, 27.

