T-lymphokine-activated killer cell-originated protein kinase (TOPK, also known as PDZ-binding kinase or PBK) plays a crucial role in cell cycle regulation and mitotic progression. Abnormal overexpression or activation of TOPK has been observed in many cancers, including colorectal cancer, triple-negative breast cancer, and melanoma, and it is associated with increased development, dissemination, and poor clinical outcomes and prognosis in cancer. Moreover, TOPK phosphorylates p38, JNK, ERK, and AKT, which are involved in many cellular functions, and participates in the activation of multiple signaling pathways related to MAPK, PI3K/PTEN/AKT, and NOTCH1; thus, the direct or indirect interactions of TOPK make it a highly attractive yet elusive target for cancer therapy. Small molecule inhibitors targeting TOPK have shown great therapeutic potential in the treatment of cancer both in vitro and in vivo, even in combination with chemotherapy or radiotherapy. Therefore, targeting TOPK could be an important approach for cancer prevention and therapy.
- TOPK
- cancer therapy
- signaling pathway
- inhibitors
1. TOPK
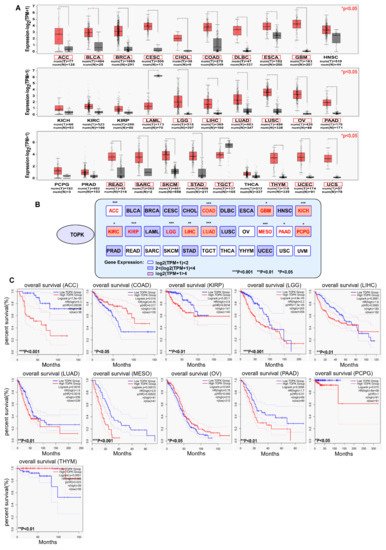
2. TOPK Expressed Highly in Cancers and Is a Prognostic Marker of Cancer
3. TOPK Promotes Proliferation
4. TOPK Promotes Tumor Dissemination
5. TOPK Regulates the Cell Cycle
6. TOPK Induces Apoptosis Resistance in Tumor Cells
7. Others Factors: Inflammation, DNA Damage, and Autophagy
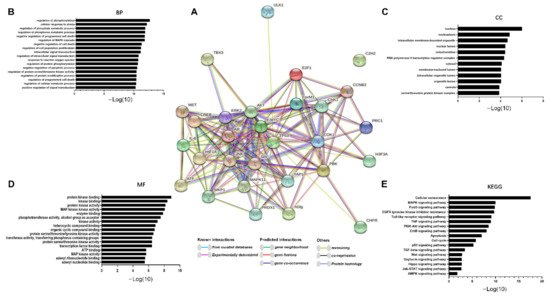
8. TOPK-Interacting Proteins Involved in Signaling Pathways
| Name | Effect Site | Function | Mechanisms | Ref |
|---|---|---|---|---|
| AKT | Phosphorylation of Ser473 | TOPK promotes AKT phosphorylation at Ser473 and decreases PTEN levels | PTEN–AKT pathway | [9] |
| AR | N/A | promotes proliferation via upregulation of PBK | A reciprocal feedback between TOPK and AR | [83] |
| CDK1 | Binds to #234–275 of TOPK, the C-terminal side of the PRC1-binding sequence | Involved in mitosis | Cell cycle arrest | [4] |
| CHK1 | CHFR ubiquitinates and regulates TOPK | Involved in mitosis | S phase arrest | [53] |
| CHFR | TOPK interacts with CHK1 | Involved in mitosis | G2 to M progression | [49] |
| c-MYC | c-Myc activate TOPK | Represses TOPK transcription | Inhibition of c-Myc- E2F1-PBK signaling that decreases cell growth and survival | [84] |
| CCNB2 | N/A | Represses TOPK transcription | PBK is essential for radioresistance by transcriptional regulation of CCNB2 | [56] |
| C2H2 | Phosphorylate C2H2 linker sequences | Regulate mitosis | Cell cycle arrest | [85] |
| CREB/ATF | Binding the PBK promoter (−312 bp) | Represses TOPK transcription | Associates with TOPK promoter | [86] |
| ERK2 | TOPK–ERK interaction | Increases ERK activity | MAPK–ERK pathway | [10] |
| E2F1 | Binding sites within the PBK promoter (−146 bp) | Represses TOPK transcription | Associates with TOPK promoter | [86] |
| FoxM1 | PBK promoter | Increases the stability and activity of TOPK | FoxM1/PBK/β-catenin signaling | [40] |
| HIF1A | N/A | Transcription of HIF1A increased by TOPK regulation | HIF-1a/snail pathway | [87] |
| H3F3A | Phosphorylation of S10 | Involved in mitosis | Cell cycle arrest | [88] |
| hDlg | Binds to the PDZ2 domain of hDlg | PBK and hDlg are phosphorylated at mitosis; phosphorylation of PBK is required for its kinase activity | PBK could link hDlg or other PDZ-containing proteins to signal transduction pathways regulating the cell cycle or cellular proliferation | [88] |
| IL-6 | N/A | Increases TOPK activity | Cell cycle arrest | [89] |
| JNK | Binds to the JNK | TOPK regulates UVB-induced JNK1 activity | TOPK enhanced the ability of JNKs to mediate H-Ras-induced cell transformation | [19] |
| LGN/GPSM2 | Thr450 | Mitosis activity | Cell cycle arrest | [55] |
| MAPK11 | TOPK-P38 interaction | Mitosis activity | p38MAPK activity and regulation of the DNA damage response | [44] |
| MET | Phosphorylates Y74 of TOPK | Increases the activity of TOPK | COX2/MET/TOPK signaling pathway | [81] |
| MKP1 | Phosphorylation of Ser-359 | TOPK directly binds with and phosphorylates MKP1 | p38 signaling pathway |
[3] |
| NF-κB | N/A | Represses TOPK transcription | ERK pathway | [22] |
| NF-κB | N/A | Activated by TOPK | Upregulation of snail/slug in TGF-β1 signaling | [90] |
| PRDX1 | Ser32 | TOPK interacts with and phosphorylates Prx1 | Induced apoptosis | [62] |
| PRC1 | T481 | TOPK interacts with and phosphorylates PRC1 | Stopping formation of mitotic spindles and spindle midzone | [4] |
| RORγ | N/A | PBK interacts with RORγ and modulates RORγ protein stability | AR-PBK- RORγ-AR pathway | [91] |
| SRC | Phosphorylation of TOPK at Y74 and Y272 | Increases the stability and activity of TOPK | TOPK–histone H3 pathway | [42] |
| TP53 | TOPK interacts with p53 through a DNA-binding domain | PBK modulation of p21 expression is dependent on p53 | Cell cycle arrest | [18] |
| TBX3 | TBX3 was activated by TOPK | TOPK upregulates T-box transcription factor TBX3 | TGF-b1/Smad signaling | [20] |
| ULK1 | Ser469, Ser495, and Ser533 | TOPK binds and phosphorylates ULK1 | Promoting ubiquitination degradation of ULK1 | [70] |
| YAP1 | N/A | Represses TOPK transcription | Hippo-YAP/TAZ pathway | [92] |
| γ-catenin | N/A | TOPK binds with γ-catenin | Src/GSK3β/STAT3 pathway | [12] |
This entry is adapted from the peer-reviewed paper 10.3390/cancers13092232