Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Wang, N. The MAMs Structure. Encyclopedia. Available online: https://encyclopedia.pub/entry/9446 (accessed on 26 June 2026).
Wang N. The MAMs Structure. Encyclopedia. Available at: https://encyclopedia.pub/entry/9446. Accessed June 26, 2026.
Wang, Nan. "The MAMs Structure" Encyclopedia, https://encyclopedia.pub/entry/9446 (accessed June 26, 2026).
Wang, N. (2021, May 10). The MAMs Structure. In Encyclopedia. https://encyclopedia.pub/entry/9446
Wang, Nan. "The MAMs Structure." Encyclopedia. Web. 10 May, 2021.
Copy Citation
Mitochondria-associated membranes (MAMs) play a vital role in the complex crosstalk between the ER and mitochondria. MAMs are known to play an important role in lipid synthesis, the regulation of Ca2+ homeostasis, the coordination of ER-mitochondrial function, and the transduction of death signals between the ER and the mitochondria.
endoplasmic reticulum
mitochondria
MAMs
Ca2+
apoptosis
1. Introduction
Apoptosis is a programmed form of cell death and can occur via three main pathways. The first pathway is the intrinsic apoptosis pathway; initiated by a range of factors, including the effects of growth factors or hormones, radiation, or cytotoxins [1]. This process involves the enhancement of pro-apoptotic signals and the weakening of anti-apoptotic signals. An imbalance in the regulation of apoptosis ultimately leads to changes in the permeability of the mitochondrial outer membrane and the release of pro-apoptotic substances from the mitochondria. These pro-apoptotic substances promote apoptosis by activating the apoptotic executive protein caspase-9, inhibiting IAPs or via the direct cleavage of DNA [2][3][4][5][6][7]. The second pathway is the extrinsic apoptosis pathway in which apoptosis is activated by the binding of specific ligands such as FasL to transmembrane receptors which contain “death domain” such as FasR. Activated death receptors then recruit adaptor proteins in the cytoplasm to assemble an apoptosis-inducing signal complex, which then activates caspase-8 to initiate apoptosis [8]. Caspase-8 can also cleave Bid to initiate the intrinsic apoptotic pathway, which plays an important role in the process of apoptotic signal amplification [9][10]. The third pathway is the perforin/granzyme pathway in which cytotoxic T cells or NK cells induce target cell apoptosis by secreting granules containing perforin or granzymes [11].
Moreover, the Bcl2 family plays a vital role in cell apoptosis. The Bcl2 family consists of 25 members, and they can be divided into pro-apoptotic proteins (Bax, Bak, etc.) and anti-apoptotic proteins (Bcl2, Bcl-XL, Mcl-1, etc.) according to their different functions. Furthermore, pro-apoptotic proteins can be divided into pro-apoptotic proteins with multiple domains and BH3-only proteins, and BH3-only proteins also can be divided into “activator” (Bim, tBid, etc.) and “sensitizer” (Bad, Bik, etc.) based on their specific mechanism of action [12]. The activated pro-apoptotic members can assemble on the outer mitochondrial membrane and change the permeability of the outer mitochondrial membrane, and promote the release of cytochrome c, AIF, Smac/Diablo and other apoptosis-inducing factors from the mitochondria [13]. Anti-apoptotic members mainly antagonize the effects of pro-apoptotic ones through protein–protein interactions to maintain the integrity of the mitochondrial outer membrane. The “activator” BH3-only members can directly activate the pro-apoptotic proteins and promote the occurrence of apoptosis, while the “sensitizer” BH3-only members can interact with the anti-apoptotic in a protein–protein interaction way to relieve the effect of the pro-apoptotic proteins [14].
As it is mentioned above, the mitochondria play a central role in the cell apoptosis, and the crosstalk between mitochondria and other organelles may impact the apoptosis process. Recent studies revealed that the communication between ER and mitochondria can influence the cell apoptosis, thus affecting the cell fate.
The ER is a key organelle that plays a crucial role in Ca2+ storage, lipid synthesis, protein folding, and assembly [15][16][17][18]. Mitochondria are the “energy factories” of eukaryotic cells, and provide energy to drive the physiological processes of cells; they also play a key role in the process of apoptosis [19][20][21]. The ER and mitochondria are independent of each other but are also closely associated in structure and function. The first spatial connection between the ER and the mitochondria was reported in the 1950s following a study of hepatocytes by transmission electron microscopy [22][23]. In 2006, an electron tomography study further confirmed the complex relationship between the ER and the mitochondria [24]. It is now believed that the mitochondrial surface juxtaposed to the ER in mammalian cells is up to 5–20% due to different cell types [25][26]. Based on this close structural connection, the ER is able to respond to a variety of stress stimuli and can transmit these stress signals to the mitochondria [27][28], thereby initiating the mitochondrial stress response. Similarly, the mitochondria can transmit signals to the ER, thus ensuring the efficient execution of compensatory responses or cell death events. Due to the special function of these precise structural associations, this biological system is usually investigated as a relatively independent sub-organelle structure referred to as a “mitochondrial-related membrane structure.”
2. The Structural Characteristics of MAMs
The structure of MAMs does not remain constant; rather, the structure of MAMs changes dynamically in response to the cell status. The width of the gap between the ER and the outer mitochondrial membrane varies from 10 to 100 nm [29][30]; the width of this gap is usually 10–15 nm at the smooth endoplasmic reticulum and 20–30 nm at the rough endoplasmic reticulum; these spatial differences may be related to the presence of ribosomes [7][31]. Different proteomic analysis of the structure of MAMs has revealed 991 [32] and 1212 [33] different proteins in MAMs [34]. Mass spectrometry analysis divided these constituent proteins into three categories: Proteins that are specifically present in MAMs; proteins that exist simultaneously in MAMs and other organelle structures; and proteins that only exist temporarily in MAMs [33]. These proteins are involved in a wide range of processes, such as structural maintenance, lipid synthesis, the regulation of Ca2+ homeostasis, mitochondrial dynamics, and apoptosis.
3. The Structure Maintenance of MAMs
There are thousands of proteins in MAMs; the roles of these proteins are known to vary widely. Some of these proteins play a tethering role in the maintenance of MAMs [31]. According to our understanding, tethering proteins should exhibit certain characteristics. For example, tethering proteins could be (1) proteins or protein complexes that directly participate in the physical connection between the ER and the mitochondria, or (2) interfering proteins or protein complexes that can directly cause changes in the width of gap, the area of contact, or the number of contact sites between the ER and the mitochondria. These proteins and protein complexes are introduced below (Figure 1).
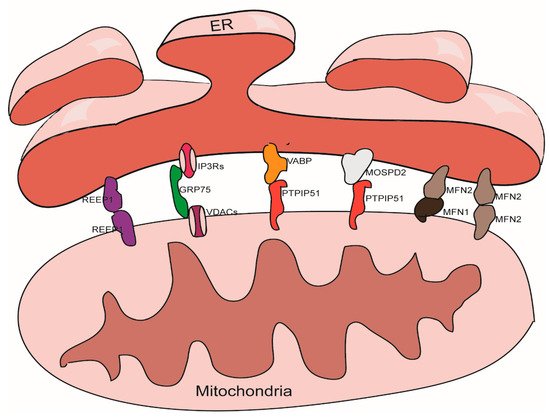
Figure 1. Tethering proteins that participate in the mitochondria-associated membranes (MAMs) structure maintenance.
3.1. The IP3Rs-Grp75-VDACs Complex
IP3Rs are important Ca2+ outflow channels on the surface of the ER and mediate the release of Ca2+ from the cavity of the ER to the cytoplasm [35][36]. VDACs are ions channel located on the outer membrane of the mitochondria; these mediate the movement of a variety of ions and metabolites in and out of mitochondria, and participate in a range of cellular activities, including apoptosis, metabolism, and the regulation of Ca2+ [37]. IP3Rs and VDACs are connected by Grp75 to maintain the structure of MAMs [38]. The overexpression of VDACs is known to enhance the connection between the ER and the mitochondria and thus improve Ca2+ flux from the ER to the mitochondria [39]; while silencing VDAC1 exhibits a reduction in the connection between Grp75 and IP3R1 indicating a reduction of ER-mitochondria interactions [40]. Cells overexpressing Grp75 showed higher number of IP3R1–VDAC1 interaction sites [41]. Silencing IP3R1 or Grp75 can also reduce the connection between VDAC1 and Grp75 or IP3R1 [42].
3.2. The VAPB-PTPIP51 Complex
VAPB is located in the membrane of the ER and participates to the activation of the IRE1/XBP1 axis in the ER unfolded protein response [43][44]. VAPB can form a complex with the outer mitochondrial membrane protein PTPIP51 and help to maintain the structure of MAMs. A mutant form of VAPB, VAPBP56S, exhibits a stronger affinity for PTPIP51, thereby promoting the transfer of Ca2+ from the ER to the mitochondria; knocking out either of these two genes can reduce the transfer of Ca2+ signals [45]. Other studies have shown that knocking down either of these two proteins will reduce the level of contacts between the ER and the mitochondria [46][47].
3.3. The Mfn1/Mfn2 Complex
In addition to being located in the outer mitochondria membrane and participating to the mitochondrial fusion [48], Mfn2 can also localize on the surface of the ER. Mfn2 participates in the structural maintenance of MAMs by forming homodimers or heterodimers with Mfn1/2 on the outer membrane of the mitochondria. The function of the Mfn1/Mfn2 complex with regards to maintaining the structure of the MAMs was first discovered in 2008 [49]; this role has also been confirmed by several other studies [50][51]. However, some studies have yielded contradictory results [52][53], it is now well established that Mfn2 plays a role in the endoplasmic reticulum stress (ERS) response; the ERS induced by knockdown of Mfn2 can tighten the association between the ER and the mitochondria [54].
3.4. The MOSPD2-PTPIP51 Complex
MOSPD2, another member of VAP family, a protein that locates on the surface of the ER membrane, plays a role in connecting the ER with other membrane structures. It can also bind with proteins containing a small VAP-interacting motif, named FFAT [two phenylalanines (FF) in an acidic track (AT)] via an MSP (Major Sperm Protein domain), such as PTPIP51 on the outer membrane of the mitochondria [55].
3.5. REEP1
REEP1 is a protein that is located in the outer membrane of the ER and the mitochondria. REEP1 helps regulate the morphology of the ER. Studies have shown that REEP1 directly connects the ER and the mitochondria through oligomerization and participates in forming the structure of MAMs. In addition, through bending ER membranes, REEP1 makes it topologically possible for the ER to wrap around the mitochondria, which helps to form MAMs [56].
3.6. Other Proteins Involved in MAMs Maintance
In addition to these tethering proteins, there are some proteins that do not directly participate in the structural maintenance of MAMs. However, these proteins do affect the structure of MAMs via protein–protein interactions (Table 1). In addition to being present in the cytoplasm, α-Synuclein can also be incorporated in MAMs [57]. α-Synuclein can promote the Ca2+ transfer from ER to mitochondria by increasing the ER and mitochondria contacts; and further study showed that the C-terminal of α-Synuclein is essential to tighten the contacts [58]. Some studies revealed that the α-Synuclein existing in MAMs results in the dis-regulation of Ca2+ and lipid metabolism, which promotes substantia nigra pars compacta neurons to die, leading to the progression of PD [59]. In addition to playing an anti-apoptotic role in cells and participating in mitochondrial dynamics, DJ-1 can still exist in the MAMs, thus enhancing the connection between the ER and the mitochondria and the crosstalk between the two organelles; this effect may be related to P53 to some extent (an antagonistic relationship) [60]. Existing studies suggest that DJ-1 can bind directly to the IP3R-Grp75-VDAC complex and affect its stability. The knockout of DJ-1 resulted in the aggregation of IP3R3 in MAMs and a reduction in the formation of the IP3Rs-Grp75-VDACs complex; it is possible that this is related to the pathophysiological process of obesity [61]. Although the precise mechanism remains obscure, it has been ascertained that DJ-1 can affect the structural stability of MAMs. This also implies that MAMs may play a role in the pathogenesis of Parkinson’s syndrome. TDP-43 and FUS are proteins that are related to ALS/FTD and can activate GSK-3b by down-regulating the phosphorylation levels of serine 9 by GSK-3b. Once activated, GSK-3b can reduce the connections between VABP and PTPIP51, thereby detaching the ER from the mitochondria [47][62]. PDK4 can directly interact with the IP3Rs-Grp75-VDACs complex in MAMs and may promote the formation of this complex by regulating phosphorylation, thus increasing the area of contacts between the ER and the mitochondria [42]. In addition to participating in the post-transcriptional modification of proteins, TG2 can also be incorporated in MAMs and act directly on Grp75 to increase the number of ER-mitochondrial contacts and thus participate in the structural maintenance of MAMs [63]. The precise function of TpMs (a type of keratin binding protein that is partly located in the mitochondria) remains unclear although data indicates that this protein can negatively regulate the ER-mitochondria connections in a Mfn2-dependent manner [64]. It is generally believed that CypD, a protein located in the mitochondrial matrix, can also be incorporated in MAMs, and directly act with the IP3Rs-Grp75-VDACs complex to regulate the stability of this complex. Inhibiting the function of CypD can down-regulate the binding of Grp75 with IP3Rs and VDACs, affecting the transfer of Ca2+ between the two organelles [65]. FUNDC1 is known for maintaining the stability of IP3R2 in MAMs by direct binding, and it enhances the level of contacts and the communication of Ca2+ between the ER and the mitochondria [66]. Presenilin-2 can also promote the connection and the transfer of Ca2+ signals between the ER and the mitochondria in the presence of Mfn-2; these findings were confirmed by overexpression and knockdown experiments, which suggested that presenilin-2 works with the Mfn1/Mfn2 complex [67]. FATE1 can reduce the level of contacts between the ER and the mitochondria and downregulate the transfer of Ca2+ with an impaired sensitivity to Ca2+-related apoptosis [68]. In addition to participating in the morphological regulation of ER, NogoB can increase the gap width of MAMs and affect their function [69]. PERK, which plays an important role in ERS, can increase the level of connectivity between the ER and the mitochondria by interacting with Mfn2, and thus promote the transduction of ERS signals to the mitochondria [70][71]. Although these proteins are not considered to be directly involved in maintaining the structure of MAMs, they still attract research attention due to their specific regulatory effects on the structure of MAMs and their involvement in the pathological processes underlying many neurodegenerative diseases.
References
- Elmore, S. Apoptosis: A review of programmed cell death. Toxicol. Pathol. 2007, 35, 495–516.
- Santucci, R.; Sinibaldi, F.; Cozza, P.; Polticelli, F.; Fiorucci, L. Cytochrome c: An extreme multifunctional protein with a key role in cell fate. Int. J. Biol. Macromol. 2019, 136, 1237–1246.
- Korga, A.; Korobowicz, E.; Dudka, J. Role of mitochondrial protein Smac/Diablo in regulation of apoptotic pathways. Pol. Merkur. Lekarski 2006, 20, 573–576.
- Vande Walle, L.; Lamkanfi, M.; Vandenabeele, P. The mitochondrial serine protease HtrA2/Omi: An overview. Cell Death Differ. 2008, 15, 453–460.
- Bano, D.; Prehn, J.H.M. Apoptosis-Inducing Factor (AIF) in Physiology and Disease: The Tale of a Repented Natural Born Killer. EBioMedicine 2018, 30, 29–37.
- Low, R.L. Mitochondrial Endonuclease G function in apoptosis and mtDNA metabolism: A historical perspective. Mitochondrion 2003, 2, 225–236.
- Larsen, B.D.; Sorensen, C.S. The caspase-activated DNase: Apoptosis and beyond. FEBS J. 2017, 284, 1160–1170.
- D’Arcy, M.S. Cell death: A review of the major forms of apoptosis, necrosis and autophagy. Cell Biol. Int. 2019, 43, 582–592.
- Kantari, C.; Walczak, H. Caspase-8 and bid: Caught in the act between death receptors and mitochondria. Biochim. Biophys. Acta 2011, 1813, 558–563.
- Huang, K.; Zhang, J.; O’Neill, K.L.; Gurumurthy, C.B.; Quadros, R.M.; Tu, Y.; Luo, X. Cleavage by Caspase 8 and Mitochondrial Membrane Association Activate the BH3-only Protein Bid during TRAIL-induced Apoptosis. J. Biol. Chem. 2016, 291, 11843–11851.
- Voskoboinik, I.; Whisstock, J.C.; Trapani, J.A. Perforin and granzymes: Function, dysfunction and human pathology. Nat. Rev. Immunol. 2015, 15, 388–400.
- Hassan, M.; Watari, H.; AbuAlmaaty, A.; Ohba, Y.; Sakuragi, N. Apoptosis and molecular targeting therapy in cancer. Biomed. Res. Int. 2014, 2014, 150845.
- Siddiqui, W.A.; Ahad, A.; Ahsan, H. The mystery of BCL2 family: Bcl-2 proteins and apoptosis: An update. Arch. Toxicol. 2015, 89, 289–317.
- Kim, H.; Rafiuddin-Shah, M.; Tu, H.C.; Jeffers, J.R.; Zambetti, G.P.; Hsieh, J.J.; Cheng, E.H. Hierarchical regulation of mitochondrion-dependent apoptosis by BCL-2 subfamilies. Nat. Cell Biol. 2006, 8, 1348–1358.
- Schwarz, D.S.; Blower, M.D. The endoplasmic reticulum: Structure, function and response to cellular signaling. Cell. Mol. Life Sci. 2016, 73, 79–94.
- Braakman, I.; Hebert, D.N. Protein folding in the endoplasmic reticulum. Cold Spring Harb. Perspect. Biol. 2013, 5, a013201.
- Quon, E.; Sere, Y.Y.; Chauhan, N.; Johansen, J.; Sullivan, D.P.; Dittman, J.S.; Rice, W.J.; Chan, R.B.; Di Paolo, G.; Beh, C.T.; et al. Endoplasmic reticulum-plasma membrane contact sites integrate sterol and phospholipid regulation. PLoS Biol. 2018, 16, e2003864.
- Islam, M.S. Calcium Signaling: From Basic to Bedside. Adv. Exp. Med. Biol. 2020, 1131, 1–6.
- Annesley, S.J.; Fisher, P.R. Mitochondria in Health and Disease. Cells 2019, 8, 680.
- Desagher, S.; Martinou, J.C. Mitochondria as the central control point of apoptosis. Trends Cell Biol. 2000, 10, 369–377.
- Galluzzi, L.; Kepp, O.; Trojel-Hansen, C.; Kroemer, G. Mitochondrial control of cellular life, stress, and death. Circ. Res. 2012, 111, 1198–1207.
- Doghman-Bouguerra, M.; Lalli, E. ER-mitochondria interactions: Both strength and weakness within cancer cells. Biochim. Biophys. Acta Mol. Cell Res. 2019, 1866, 650–662.
- Bernhard, W.; Rouiller, C. Close topographical relationship between mitochondria and ergastoplasm of liver cells in a definite phase of cellular activity. J. Biophys. Biochem. Cytol. 1956, 2, 73–78.
- Mannella, C.A. The relevance of mitochondrial membrane topology to mitochondrial function. Biochim. Biophys. Acta 2006, 1762, 140–147.
- Rizzuto, R.; Pinton, P.; Carrington, W.; Fay, F.S.; Fogarty, K.E.; Lifshitz, L.M.; Tuft, R.A.; Pozzan, T. Close contacts with the endoplasmic reticulum as determinants of mitochondrial Ca2+ responses. Science 1998, 280, 1763–1766.
- Marchi, S.; Patergnani, S.; Missiroli, S.; Morciano, G.; Rimessi, A.; Wieckowski, M.R.; Giorgi, C.; Pinton, P. Mitochondrial and endoplasmic reticulum calcium homeostasis and cell death. Cell Calcium 2018, 69, 62–72.
- Iurlaro, R.; Munoz-Pinedo, C. Cell death induced by endoplasmic reticulum stress. FEBS J. 2016, 283, 2640–2652.
- Gardner, B.M.; Pincus, D.; Gotthardt, K.; Gallagher, C.M.; Walter, P. Endoplasmic reticulum stress sensing in the unfolded protein response. Cold Spring Harb. Perspect. Biol. 2013, 5, a013169.
- Giacomello, M.; Pellegrini, L. The coming of age of the mitochondria-ER contact: A matter of thickness. Cell Death Differ. 2016, 23, 1417–1427.
- Filadi, R.; Theurey, P.; Pizzo, P. The endoplasmic reticulum-mitochondria coupling in health and disease: Molecules, functions and significance. Cell Calcium 2017, 62, 1–15.
- Csordas, G.; Renken, C.; Varnai, P.; Walter, L.; Weaver, D.; Buttle, K.F.; Balla, T.; Mannella, C.A.; Hajnoczky, G. Structural and functional features and significance of the physical linkage between ER and mitochondria. J. Cell Biol. 2006, 174, 915–921.
- Zhang, A.; Williamson, C.D.; Wong, D.S.; Bullough, M.D.; Brown, K.J.; Hathout, Y.; Colberg-Poley, A.M. Quantitative proteomic analyses of human cytomegalovirus-induced restructuring of endoplasmic reticulum-mitochondrial contacts at late times of infection. Mol. Cell. Proteom. 2011, 10, M111-009936.
- Poston, C.N.; Krishnan, S.C.; Bazemore-Walker, C.R. In-depth proteomic analysis of mammalian mitochondria-associated membranes (MAM). J. Proteom. 2013, 79, 219–230.
- Janikiewicz, J.; Szymanski, J.; Malinska, D.; Patalas-Krawczyk, P.; Michalska, B.; Duszynski, J.; Giorgi, C.; Bonora, M.; Dobrzyn, A.; Wieckowski, M.R. Mitochondria-associated membranes in aging and senescence: Structure, function, and dynamics. Cell Death Dis. 2018, 9, 332.
- Parys, J.B.; Vervliet, T. New Insights in the IP3 Receptor and Its Regulation. Adv. Exp. Med. Biol. 2020, 1131, 243–270.
- Tada, M.; Nishizawa, M.; Onodera, O. Roles of inositol 1,4,5-trisphosphate receptors in spinocerebellar ataxias. Neurochem. Int. 2016, 94, 1–8.
- Mazure, N.M. VDAC in cancer. Biochim. Biophys. Acta Bioenerg. 2017, 1858, 665–673.
- Szabadkai, G.; Bianchi, K.; Varnai, P.; De Stefani, D.; Wieckowski, M.R.; Cavagna, D.; Nagy, A.I.; Balla, T.; Rizzuto, R. Chaperone-mediated coupling of endoplasmic reticulum and mitochondrial Ca2+ channels. J. Cell Biol. 2006, 175, 901–911.
- Rapizzi, E.; Pinton, P.; Szabadkai, G.; Wieckowski, M.R.; Vandecasteele, G.; Baird, G.; Tuft, R.A.; Fogarty, K.E.; Rizzuto, R. Recombinant expression of the voltage-dependent anion channel enhances the transfer of Ca2+ microdomains to mitochondria. J. Cell Biol. 2002, 159, 613–624.
- Tubbs, E.; Theurey, P.; Vial, G.; Bendridi, N.; Bravard, A.; Chauvin, M.A.; Ji-Cao, J.; Zoulim, F.; Bartosch, B.; Ovize, M.; et al. Mitochondria-associated endoplasmic reticulum membrane (MAM) integrity is required for insulin signaling and is implicated in hepatic insulin resistance. Diabetes 2014, 63, 3279–3294.
- Honrath, B.; Metz, I.; Bendridi, N.; Rieusset, J.; Culmsee, C.; Dolga, A.M. Glucose-regulated protein 75 determines ER-mitochondrial coupling and sensitivity to oxidative stress in neuronal cells. Cell Death Discov. 2017, 3, 17076.
- Thoudam, T.; Ha, C.M.; Leem, J.; Chanda, D.; Park, J.S.; Kim, H.J.; Jeon, J.H.; Choi, Y.K.; Liangpunsakul, S.; Huh, Y.H.; et al. PDK4 Augments ER-Mitochondria Contact to Dampen Skeletal Muscle Insulin Signaling during Obesity. Diabetes 2019, 68, 571–586.
- Vinay Kumar, C.; Kumar, K.M.; Swetha, R.; Ramaiah, S.; Anbarasu, A. Protein aggregation due to nsSNP resulting in P56S VABP protein is associated with amyotrophic lateral sclerosis. J. Theor. Biol. 2014, 354, 72–80.
- Kanekura, K.; Nishimoto, I.; Aiso, S.; Matsuoka, M. Characterization of amyotrophic lateral sclerosis-linked P56S mutation of vesicle-associated membrane protein-associated protein B (VAPB/ALS8). J. Biol. Chem. 2006, 281, 30223–30233.
- De Vos, K.J.; Morotz, G.M.; Stoica, R.; Tudor, E.L.; Lau, K.F.; Ackerley, S.; Warley, A.; Shaw, C.E.; Miller, C.C. VAPB interacts with the mitochondrial protein PTPIP51 to regulate calcium homeostasis. Hum. Mol. Genet. 2012, 21, 1299–1311.
- Stoica, R.; De Vos, K.J.; Paillusson, S.; Mueller, S.; Sancho, R.M.; Lau, K.F.; Vizcay-Barrena, G.; Lin, W.L.; Xu, Y.F.; Lewis, J.; et al. ER-mitochondria associations are regulated by the VAPB-PTPIP51 interaction and are disrupted by ALS/FTD-associated TDP-43. Nat. Commun. 2014, 5, 3996.
- Qiao, X.; Jia, S.; Ye, J.; Fang, X.; Zhang, C.; Cao, Y.; Xu, C.; Zhao, L.; Zhu, Y.; Wang, L.; et al. PTPIP51 regulates mouse cardiac ischemia/reperfusion through mediating the mitochondria-SR junction. Sci. Rep. 2017, 7, 45379.
- Formosa, L.E.; Ryan, M.T. Mitochondrial fusion: Reaching the end of mitofusin’s tether. J. Cell Biol. 2016, 215, 597–598.
- de Brito, O.M.; Scorrano, L. Mitofusin 2 tethers endoplasmic reticulum to mitochondria. Nature 2008, 456, 605–610.
- Alford, S.C.; Ding, Y.; Simmen, T.; Campbell, R.E. Dimerization-dependent green and yellow fluorescent proteins. ACS Synth. Biol. 2012, 1, 569–575.
- Naon, D.; Zaninello, M.; Giacomello, M.; Varanita, T.; Grespi, F.; Lakshminaranayan, S.; Serafini, A.; Semenzato, M.; Herkenne, S.; Hernandez-Alvarez, M.I.; et al. Critical reappraisal confirms that Mitofusin 2 is an endoplasmic reticulum-mitochondria tether. Proc. Natl. Acad. Sci. USA 2016, 113, 11249–11254.
- Cosson, P.; Marchetti, A.; Ravazzola, M.; Orci, L. Mitofusin-2 independent juxtaposition of endoplasmic reticulum and mitochondria: An ultrastructural study. PLoS ONE 2012, 7, e46293.
- Filadi, R.; Greotti, E.; Turacchio, G.; Luini, A.; Pozzan, T.; Pizzo, P. Mitofusin 2 ablation increases endoplasmic reticulum-mitochondria coupling. Proc. Natl. Acad. Sci. USA 2015, 112, E2174–E2181.
- Herrera-Cruz, M.S.; Simmen, T. Cancer: Untethering Mitochondria from the Endoplasmic Reticulum? Front. Oncol. 2017, 7, 105.
- Di Mattia, T.; Wilhelm, L.P.; Ikhlef, S.; Wendling, C.; Spehner, D.; Nomine, Y.; Giordano, F.; Mathelin, C.; Drin, G.; Tomasetto, C.; et al. Identification of MOSPD2, a novel scaffold for endoplasmic reticulum membrane contact sites. EMBO Rep. 2018, 19.
- Lim, Y.; Cho, I.T.; Schoel, L.J.; Cho, G.; Golden, J.A. Hereditary spastic paraplegia-linked REEP1 modulates endoplasmic reticulum/mitochondria contacts. Ann. Neurol. 2015, 78, 679–696.
- Guardia-Laguarta, C.; Area-Gomez, E.; Rub, C.; Liu, Y.; Magrane, J.; Becker, D.; Voos, W.; Schon, E.A.; Przedborski, S. alpha-Synuclein is localized to mitochondria-associated ER membranes. J. Neurosci. 2014, 34, 249–259.
- Cali, T.; Ottolini, D.; Negro, A.; Brini, M. alpha-Synuclein controls mitochondrial calcium homeostasis by enhancing endoplasmic reticulum-mitochondria interactions. J. Biol. Chem. 2012, 287, 17914–17929.
- Gomez-Suaga, P.; Bravo-San Pedro, J.M.; Gonzalez-Polo, R.A.; Fuentes, J.M.; Niso-Santano, M. ER-mitochondria signaling in Parkinson’s disease. Cell Death Dis. 2018, 9, 337.
- Ottolini, D.; Cali, T.; Negro, A.; Brini, M. The Parkinson disease-related protein DJ-1 counteracts mitochondrial impairment induced by the tumour suppressor protein p53 by enhancing endoplasmic reticulum-mitochondria tethering. Hum. Mol. Genet. 2013, 22, 2152–2168.
- Liu, Y.; Ma, X.; Fujioka, H.; Liu, J.; Chen, S.; Zhu, X. DJ-1 regulates the integrity and function of ER-mitochondria association through interaction with IP3R3-Grp75-VDAC1. Proc. Natl. Acad. Sci. USA 2019, 116, 25322–25328.
- Stoica, R.; Paillusson, S.; Gomez-Suaga, P.; Mitchell, J.C.; Lau, D.H.; Gray, E.H.; Sancho, R.M.; Vizcay-Barrena, G.; De Vos, K.J.; Shaw, C.E.; et al. ALS/FTD-associated FUS activates GSK-3beta to disrupt the VAPB-PTPIP51 interaction and ER-mitochondria associations. EMBO Rep. 2016, 17, 1326–1342.
- D’Eletto, M.; Rossin, F.; Occhigrossi, L.; Farrace, M.G.; Faccenda, D.; Desai, R.; Marchi, S.; Refolo, G.; Falasca, L.; Antonioli, M.; et al. Transglutaminase Type 2 Regulates ER-Mitochondria Contact Sites by Interacting with GRP75. Cell Rep. 2018, 25, 3573–3581 e3574.
- Cerqua, C.; Anesti, V.; Pyakurel, A.; Liu, D.; Naon, D.; Wiche, G.; Baffa, R.; Dimmer, K.S.; Scorrano, L. Trichoplein/mitostatin regulates endoplasmic reticulum-mitochondria juxtaposition. EMBO Rep. 2010, 11, 854–860.
- Paillard, M.; Tubbs, E.; Thiebaut, P.A.; Gomez, L.; Fauconnier, J.; Da Silva, C.C.; Teixeira, G.; Mewton, N.; Belaidi, E.; Durand, A.; et al. Depressing mitochondria-reticulum interactions protects cardiomyocytes from lethal hypoxia-reoxygenation injury. Circulation 2013, 128, 1555–1565.
- Wu, S.; Lu, Q.; Wang, Q.; Ding, Y.; Ma, Z.; Mao, X.; Huang, K.; Xie, Z.; Zou, M.H. Binding of FUN14 Domain Containing 1 With Inositol 1,4,5-Trisphosphate Receptor in Mitochondria-Associated Endoplasmic Reticulum Membranes Maintains Mitochondrial Dynamics and Function in Hearts In Vivo. Circulation 2017, 136, 2248–2266.
- Filadi, R.; Greotti, E.; Turacchio, G.; Luini, A.; Pozzan, T.; Pizzo, P. Presenilin 2 Modulates Endoplasmic Reticulum-Mitochondria Coupling by Tuning the Antagonistic Effect of Mitofusin 2. Cell Rep. 2016, 15, 2226–2238.
- Doghman-Bouguerra, M.; Granatiero, V.; Sbiera, S.; Sbiera, I.; Lacas-Gervais, S.; Brau, F.; Fassnacht, M.; Rizzuto, R.; Lalli, E. FATE1 antagonizes calcium- and drug-induced apoptosis by uncoupling ER and mitochondria. EMBO Rep. 2016, 17, 1264–1280.
- Sutendra, G.; Dromparis, P.; Wright, P.; Bonnet, S.; Haromy, A.; Hao, Z.; McMurtry, M.S.; Michalak, M.; Vance, J.E.; Sessa, W.C.; et al. The role of Nogo and the mitochondria-endoplasmic reticulum unit in pulmonary hypertension. Sci. Transl. Med. 2011, 3, 88ra55.
- Munoz, J.P.; Ivanova, S.; Sanchez-Wandelmer, J.; Martinez-Cristobal, P.; Noguera, E.; Sancho, A.; Diaz-Ramos, A.; Hernandez-Alvarez, M.I.; Sebastian, D.; Mauvezin, C.; et al. Mfn2 modulates the UPR and mitochondrial function via repression of PERK. EMBO J. 2013, 32, 2348–2361.
- van Vliet, A.R.; Agostinis, P. When under pressure, get closer: PERKing up membrane contact sites during ER stress. Biochem. Soc. Trans. 2016, 44, 499–504.
More
Information
Subjects:
Biology
Contributor
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
992
Revisions:
2 times
(View History)
Update Date:
10 May 2021
Table of Contents


Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No