 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Kiyotaka Itcho | + 3245 word(s) | 3245 | 2021-04-23 05:40:52 | | | |
| 2 | Conner Chen | Meta information modification | 3245 | 2021-05-07 03:17:02 | | |
Video Upload Options
Primary aldosteronism (PA) is the most common form of secondary hypertension, with a prevalence of 5–10% among patients with hypertension. PA is mainly classified into two subtypes: aldosterone-producing adenoma (APA) and bilateral idiopathic hyperaldosteronism. Recent developments in genetic analysis have facilitated the discovery of mutations in KCNJ5, ATP1A1, ATP2B3, CACNA1D, CACNA1H, CLCN2, and CTNNB1 in sporadic or familial forms of PA in the last decade. These findings have greatly advanced our understanding of the mechanism of excess aldosterone synthesis, particularly in APA. Most of the causative genes encode ion channels or pumps, and their mutations lead to depolarization of the cell membrane due to impairment of ion transport. Depolarization activates voltage-gated Ca2+ channels and intracellular calcium signaling and promotes the transcription of aldosterone synthase, resulting in overproduction of aldosterone.
1. Primary aldosteronism
Aldosterone is synthesized in the adrenal cortex and plays an essential role in regulating blood pressure by promoting sodium reabsorption in the kidney. Primary aldosteronism (PA), which is a disorder of excess aldosterone secretion, is the most common form of secondary hypertension, with a prevalence of 5–10% among patients with hypertension [1]. The risk of cardiometabolic and renal disease is higher in PA patients than in essential hypertension patients; thus, early diagnosis and appropriate treatment of PA are important for reducing its complications [2][3][4][5]. PA is mainly classified into two subtypes: aldosterone-producing adenoma (APA) and bilateral idiopathic hyperaldosteronism (BHA). Although the etiology of PA has long remained unclear, recent developments in genetic analysis, including next-generation sequencing (NGS), have expanded our understanding of the genetic and molecular mechanisms of PA in the last decade. Exome sequencing discovered somatic mutations in KCNJ5, ATP1A1, ATP2B3, CACNA1D, CACNA1H, CLCN2, and CTNNB1 in APA [6][7][8][9][10][11][12][13]. Most of the causative genes encode ion channels or pumps, and their mutations lead to depolarization of the cell membrane due to impairment of ion transport. Depolarization activates voltage-gated Ca2+ channels and intracellular calcium signaling and promotes the transcription of aldosterone synthase (CYP11B2), resulting in overproduction of aldosterone (Figure 1). Furthermore, some key molecules such as VSNL1, CALN1, GSTA1, NPNT, and CLGN have been detected in APA, and their functions in aldosterone production have been elucidated [14][15][16][17][18]. Epigenetic regulation of CYP11B2 has also been indicated in APA [19][20][21][22].
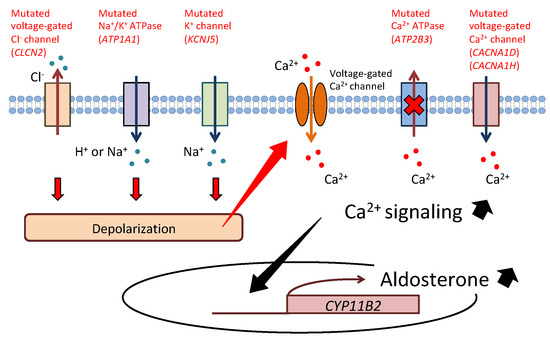
Figure 1. Cellular mechanism of aldosterone synthesis in aldosterone-producing adenoma. Mutations of KCNJ5, ATP1A1, and CLCN2 lead to depolarization of the cell membrane due to impairment of ion transport. Depolarization activates voltage-gated Ca2+ channels and increases intracellular Ca2+ levels. Conversely, mutations of CACNA1D and CACNA1H directly cause an increase in Ca2+ conductance. ATP2B3 mutation reduces Ca2+ export from the cell. Activated calcium signaling promotes transcription of aldosterone synthase (CYP11B2), resulting in overproduction of aldosterone.
Familial hyperaldosteronism (FH) has also been reported as a rare cause of PA. There are four forms of FH (FH type 1 to type 4). Although it is rare, the study of FH was preferred as an approach to understand the pathophysiology of PA due to its heritability. The first report of FH was the case of a father and a son presenting the symptoms of PA in 1966, which was corrected by glucocorticoid treatment [23]. Thus, this form of PA is called glucocorticoid-remediable aldosteronism (GRA) or FH type 1. In 1992, linkage analysis revealed that the molecular etiology of GRA was a chimeric gene composed of the promoter of 11β-hydroxylase (CYP11B1) fused with the coding region of CYP11B2, resulting in aldosterone overproduction regulated by ACTH [24]. The chimeric CYP11B1/CYP11B2 gene was not identified in APA [25], whereas some causative genes, including KCNJ5, CLCN2, and CACNA1H, have been discovered in the other forms of FH [6][10][11][12].
2. KCNJ5
In 2011, Choi et al. analyzed 22 cases of APA using whole-exome sequencing and identified two recurrent somatic mutations of KCNJ5 (G151R and L168R) [6]. KCNJ5 encodes the G protein-coupled inwardly rectifying K+ channel (GIRK4), which belongs to GIRK family members (GIRK1 to GIRK4). GIRK4, which consists of two membrane-spanning domains, one pore-forming region between the two transmembrane domains, and intracellular N and C termini, forms a channel as a homotetramer or heterotetramer with GIRK1. Both substitutions are located near the channel’s ion-selective filter and cause depolarization of the cell membrane due to the loss of ion selectivity of the K+ channel and the increased intracellular influx of Na+. The authors proposed that activated voltage-gated Ca2+ channels resulting from these mutations promote autonomous secretion of aldosterone and cell proliferation. In subsequent studies with adrenocortical carcinoma cell lines, introduction of the KCNJ5 mutation promoted aldosterone synthesis via depolarization of the cell membrane, allowing sodium and calcium influx into the cell [26][27][28][29]. Mutated KCNJ5 also increased the expression of CYP11B2 with its transcription factors nuclear receptor related 1 (Nurr1) and activating transcription factor 2 (ATF2), and these stimulatory effects were inhibited by Ca2+ channel blockers [26][27][30]. Moreover, molecules related to calcium signaling, such as VSNL1 and CALN1, are highly expressed in APA, and they have important roles in aldosterone production [14][15][31]. These results show that increased CYP11B2 expression is mediated by the Ca2+/calmodulin cascade. The relationship between KCNJ5 mutation and cell proliferation is still controversial, and the difference in KCNJ5 mutation modulation levels may influence adrenal cell growth [26][32][33]. Several other KCNJ5 mutations such as E145Q, I157del, and T158A have been reported, although G151R and L168R are the most frequent [8][29][34][35][36][37][38][39][40][41][42][43][44][45].
KCNJ5 is the most commonly mutated somatic gene in Asians, Europeans, and Americans with APA [38][41][45]. In a report of 474 APA cases from the European Network for the Study of Adrenal Tumors (ENS@T), KCNJ5 mutation was found in 38% of cases [45]. In White Americans and African Americans, KCNJ5 mutation was found in 43% and 34% of cases, respectively [37][42]. Conversely, reports from East Asia have shown that nearly 70% of APA patients have a KCNJ5 mutation, with an ethnic difference [41][43][46][47][48][49][50]. A meta-analysis showed that APA patients with KCNJ5 mutation have phenotypic features of higher plasma aldosterone levels, young age, female sex, and larger tumor size [51]. Subclinical hypercortisolism is sometimes accompanied by APA; aldosterone and cortisol co-producing adenoma has also been reported in KCNJ5-mutated APA [52]. However, a recent prospective study showed that subclinical hypercortisolism was common in APA without KCNJ5 mutation or with a relatively larger tumor size [53]. Cardiovascular complications in APA patients with KCNJ5 mutations also have been evaluated in some studies. In KCNJ5-mutated APA patients, higher left ventricular mass index (LVMI) and plasma aldosterone levels were reported than in those without KCNJ5 mutation [54]. Another group reported that the KCNJ5-mutated group significantly improved LVMI after surgery [55]. A recent study also showed that APA patients with KCNJ5 mutations had higher LVMI and inappropriately excessive LVMI (ieLVMI), as well as a greater regression of LVMI and ieLVMI after adrenalectomy, in comparison to those without KCNJ5 mutations in a propensity-score-matched cohort [56]. These results indicate KCNJ5 mutation is associated with left ventricular remodeling and diastolic function. KCNJ5 mutation was also reported to be a predictor of hypertension remission after adrenalectomy for APA [43][57]. On the other hand, subclinical hypercortisolism in patients with APA was indicated to be associated with a lower clinical complete success rate after adrenalectomy [53].
The adrenal cortex comprises three morphologically and functionally distinct layers: zona glomerulosa (ZG), zona fasciculata (ZF), and zona reticularis (ZR). Although the expressions of steroid enzymes are zone-specific, the histological features of APA are heterogeneous [58]. CYP11B2 is specifically expressed in ZG, and 17α-hydroxylase/17,20-lyase (CYP17A1) is expressed in ZF and ZR in the normal adult adrenal gland; however, APA with a KCNJ5 mutation typically has predominant clear cells (ZF-like cells) [59], and expression of both CYP11B2 and CYP17A1 is found within the same tumor [60][61]. Plasma levels of the hybrid steroids 18-oxocortisol and 18-hydroxycortisol have been reported to be higher in APA patients, particularly in KCNJ5-mutated APA [62], which could be explained by its ZF-significant phenotype (Figure 2.) [63]. Thus, steroids have been indicated as clinical biomarkers, and steroid profiling can be utilized for differentiating subtypes of PA [64][65][66][67].
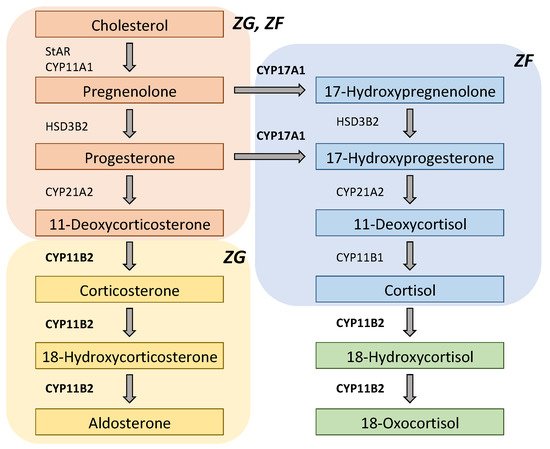
Figure 2. Scheme of steroidogenic pathways for aldosterone, 18-oxocortisol, and 18-hydroxycortisol. Both CYP11B2 (aldosterone synthase) and CYP17A1 (17α-hydroxylase/17,20-lyase) are required to synthesize 18-oxocortisol and 18-hydroxycortisol. Thus, plasma levels of 18-oxocortisol and 18-hydroxycortisol are likely to be higher in patients with KCNJ5-mutated aldosterone-producing adenoma (APA), while they are very low in normal adults. CYP11A1: cytochrome P450 cholesterol side-chain cleavage; CYP11B1: 11β-hydroxylase; CYP21A2: 21-hydroxylase; HSD3B2: 3β-hydroxysteroid dehydrogenase type 2; StAR: steroidogenic acute regulatory protein; ZF: zona fasciculata; ZG: zona glomerulosa.
Germline mutation in KCNJ5 also has been identified in FH. In 2008, Geller et al. reported the case of a father and two daughters with a new form of PA [68]. They showed early-onset PA and marked adrenocortical hyperplasia, which did not respond to medical therapy and led to bilateral adrenalectomy. Choi et al. genetically analyzed this family and discovered germline KCNJ5 mutation responsible for the disease, which was later classified as FH type 3 [6]. Since then, various phenotypes of FH type 3 depending on genotype have been reported; T158A, I157S, E145Q, and G151R are reported to have severe early-onset PA with bilateral adrenal hyperplasia, requiring bilateral adrenalectomy [6][69][70][71]. On the other hand, G151E and Y152C are associated with mild PA with no adrenal abnormalities on computed tomography (CT) scan and can be controlled by mineralocorticoid receptor antagonist (MRA) [71][72][73]. In vitro study demonstrated that transduction of KCNJ5 G151E leads to profoundly large Na+ conductance compared with other mutations, leading to Na+-influx-dependent cell lethality [71][72]. Therefore, it is suggested that these marked alterations of channel function prevent the development of adrenal hyperplasia, resulting in a mild clinical phenotype. However, there was a report of the early-onset PA with de novo KCNJ5 G151R germline mutation and no adrenal enlargement whose symptoms were successfully controlled by MRA, indicating that diverse clinical phenotype in FH type 3 cannot be defined solely by KCNJ5 genotype [74]. In addition, two cases of early-onset PA possibly caused by mosaicism for KCNJ5 mutations were reported [75][76].
3. ATP1A1
Beuschlein et al. identified a somatic mutation in ATP1A1 in 16/308 (5.2%) APAs [7], and Azizan et al. found it in 2 of 10 ZG-like APAs without KCNJ5 mutation [8]. In contrast to KCNJ5-mutated APA, APA with ATP1A1 mutation is more commonly found in males and has histological features of predominant ZG-like cells [7][8]. ATP1A1 encodes the alpha 1 subunit of Na+/K+ ATPase, which transports three Na+ ions in exchange for two K+ ions. The alpha subunit is composed of 10 transmembrane domains (M1–M10) with intracellular N and C termini. Several somatic mutations such as G99R, L104R, V332G, and EETA963S were identified in the M1, M4, and M9 domains [7][8][35]. Mutations in the M1 and M4 domains, which result in alteration of K+ binding and loss of pump activity, lead to depolarization of the cell membrane and autonomous secretion of aldosterone [7]. Mutations in the M9 domain affect a supposed Na+-specific site, resulting in loss of pump activity [8]. These mutations were suggested to lead to abnormal H+ or Na+ leakage current, which is a similar mechanism to that of the KCNJ5 mutation [8]. However, in vitro study using adrenocortical cells demonstrated that mutations in ATP1A1 induce depolarization of the cell membrane and intracellular acidification due to H+ leak, but not an overt increase in intracellular Ca2+ [77]. The specific mechanism of this acidification in autonomous aldosterone production has not been clarified.
The frequency of ATP1A1 mutation determined through Sanger sequencing performed on whole tumor sample DNA was not as high as that of KCNJ5 reported previously. However, a recently developed sequencing method using targeted NGS performed on DNA extracted from formalin-fixed paraffin-embedded tissues expressing CYP11B2 in immunohistochemistry (IHC) has enabled the more frequent detection of somatic mutations in APA [37]. The CYP11B2 IHC-guided targeted NGS approach identified 5.0–17% of ATP1A1 mutations in APA cases [37][42][78][79], whereas the frequency of ATP1A1 mutations was 2.4–8.2% using conventional methods [7][35][38][41][45]. There are few reports of specific clinical characteristics of APA patients with non-KCNJ5 mutation; one report showed that APA patients with ATPase mutation tended to have more severe hyperaldosteronism compared to those with wild type, although the sample size was small [80].
4. ATP2B3
ATP2B3 encodes the plasma membrane Ca2+ ATPase type 3 (PMCA3), which exports calcium ions from the cytoplasm. Beuschlein et al. reported somatic mutation of ATP2B3 along with that of ATP1A1 in APA [7]. PMCA3 is also composed of 10 transmembrane domains (M1–M10) with intracellular N and C termini. Most of the mutations identified in APA are deletion mutations located in the specific region of the M4 domain, which is involved in Ca2+ binding and ion gating [7][37][38][41][42][45][78][79][81]. This mutation is presumed to cause a major distortion of the Ca2+ binding site, impairing the clearance of cytoplasmic Ca2+ ions. Subsequent in vitro studies have demonstrated that ATP2B3 mutation promotes aldosterone production by two different mechanisms: (1) reduction of Ca2+ export due to the loss of pump function increases resting Ca2+ activity and (2) influx of Na+ caused by gain of cation permeability leads to depolarization and activates voltage-gated Ca2+ channels [82]. The frequency of ATP2B3 mutation is relatively low, accounting for 0.6–10% of APA cases [7][35][37][38][41][42][45][78][79]. ATP2B3 mutation was also frequently found in APA mainly composed of ZG-like cells [58][70][83]. However, a recent study using a quantitative histological analytical approach with digital imaging software showed that ATP2B3-mutated APA tended to have clear cell dominant features [61].
5. CACNA1D
Scholl et al. identified five somatic CACNA1D mutations (G403R and I770M) among 43 APAs without KCNJ5 mutation [9]. CACNA1D encodes a calcium channel voltage-dependent L-type alpha-1D subunit, which contains four repeated domains (I–IV), each with six transmembrane segments (S1–S6). These altered residues locate in the S6 segments lining the channel pore and induce a shift in voltage-dependent gating to a more negative voltage, leading to an increase in intracellular Ca2+ levels [9]. However, subsequent studies have shown that somatic mutations in CACNA1D are found throughout the gene in APA [84]. Azizan et al. also reported somatic CACNA1D mutations in ZG-like APA at the same time [8]. They also reported that CACNA1D mutations were associated with small tumor size, but this association was not found in a recent study using the CYP11B2 IHC-guided targeted NGS approach [79]. The CYP11B2 IHC-guided targeted NGS approach identified a large number of CACNA1D mutations (14–42%) [37][42][78][79] compared to conventional methods (0.6–10.3%) [38][41][45]. Moreover, CACNA1D mutations are most prevalent (42%), followed by KCNJ5 mutations, in African American patients with APA [42].
Scholl et al. also reported de novo germline CACNA1D mutations (G403D and I770M) in two children featuring early-onset PA with seizures and neurologic abnormalities (PASNA). Although several cases of neurodevelopmental disease with CACNA1D de novo germline mutations have been reported, only four cases presenting early-onset PA have been described to date [9][85][86]. Treatment with calcium channel blockers (amlodipine and nifedipine) normalized blood pressure in two of these cases [9][86], and CT scan showed no adrenal abnormality in one case [9].
6. CTNNB1
CTNNB1 encodes β-catenin, and its mutation induces constitutive activation of Wnt/β-catenin signaling. Although Wnt/β-catenin signaling plays a crucial role in normal development and maintenance of the adrenal cortex [87], activated Wnt/β-catenin signaling is also observed in APA [88][89]. In addition to ion channels and ATPases, mutations in CTNNB1 have been reported in APA with 0–5.1% frequency [13][37][42][78][79][90]. The extracellular matrix gene NPNT, which is downstream of the Wnt/β-catenin signaling pathway, is upregulated in ZG-like APA, especially with CTNNB1 mutation. NPNT over-expression increases aldosterone production in adrenal cells [17]. CTNNB1 mutation has also been found in other adrenocortical adenomas and adrenocortical carcinomas [91]. A previous study showed that transgenic mice with constitutive β-catenin activation in adrenal tumors develop hyperaldosteronism and malignancy [92]. Taken together, these results suggest that CTNNB1 mutations stimulate ZG cell proliferation and Wnt/β-catenin activation participates in aldosterone production. APA with CTNNB1 mutation is more common in females and has variable histological features [13][90]. A higher risk of residual hypertension after adrenalectomy in patients with CTNNB1-mutated APA was shown in one report [90]. Clinical and histological features of APA harboring each somatic mutation are summarized in Table 1.
Table 1. Clinical and histological features of APA harboring each somatic mutation.
| Gene | Clinical Characteristics | Histological Features |
|---|---|---|
| KCNJ5 | More common in Asians More often female Diagnosed at younger age Larger tumor size Higher plasma levels of aldosterone, 18-oxocortisol, and 18-hydroxycortisol More likely to have hypertension remission after adrenalectomy |
Clear cell dominant (ZF-like) |
| ATP1A1 | More often male Smaller tumor size |
Compact cell dominant (ZG-like) |
| ATP2B3 | More often male Smaller tumor size |
Compact cell dominant (ZG-like) |
| CACNA1D | More common in African Americans More often male Smaller tumor size |
Compact cell dominant (ZG-like) |
| CTNNB1 | More often female Higher risk of post adrenalectomy residual hypertension |
Variable |
7. CLCN2
In 1991, Gordon et al. reported six relatives who presented with APA or BHA unresponsive to glucocorticoids [93]. Several other familial cases were reported by the same group, which was defined as FH type 2 [94]. The cause of FH type 2 had been unknown for a long time; in 2018, Scholl et al. identified CLCN2 R172Q germline mutation as the cause of FH type 2 by performing exome sequencing on these families [11]. They further analyzed 80 other young-onset PAs without known mutations and reported several CLCN2 germline mutations with a frequency of 9.9% [11]. At the same time, Fernandes-Rosa et al. also analyzed 12 young-onset PAs and discovered CLCN2 G24D de novo germline mutation [12]. CLCN2 encodes the inwardly rectifying chloride channel ClC2, which is expressed in many tissues, including the adrenal glands. These mutations cause depolarization of the plasma membrane by promoting efflux of Cl– ions through gain of function and activation of CYP11B2 transcription from voltage-gated Ca2+ channel activity [11][12]. The morphology of the adrenal glands varied from normal to unilateral nodules on CT scan, but aldosterone production was bilateral in the three cases that underwent adrenal venous sampling [11]. Recently, somatic mutations of CLCN2 were reported in sporadic APA, but the frequency is rare [95][96].
8. CACNA1H
In 2015, Scholl et al. performed exome sequencing in 40 hypertensive patients who developed PA before the age of 10 years and identified the CACNA1H M1549V germline mutation in five patients, which was classified as FH type 4 [10]. This mutation occurred de novo in two patients and was inherited in the remaining three [10]. CACNA1H encodes a voltage-dependent Ca2+ channel T-type alpha-1H subunit (Cav3.2), which is the second most highly expressed calcium channel alpha subunit after CACNA1D in the human adrenal cortex [9]. This mutation reduces the normal inactivation of Cav3.2 compared with wild type and also activates the channel with less depolarization, causing intracellular Ca2+ influx, which is a similar mechanism to the CACNA1D mutation [10]. They did not show neurodevelopmental symptoms seen in PASNA and adrenal hyperplasia on CT scan, although one sporadic APA case with multiplex developmental disorder and germline CACNA1H mutation was reported [10][97]. The clinical and molecular characteristics of FH are summarized in Table 2. In addition, somatic CACNA1H mutations were also reported in sporadic APAs without known mutations using the CYP11B2 IHC-guided sequencing approach [78][98].
Table 2. Clinical and molecular characteristics of familial hyperaldosteronism (FH).
| Genetic Variant | Molecular Mechanism | Clinical Characteristics | |
|---|---|---|---|
| Type 1 | CYP11B1/CYP11B2 chimeric gene |
ACTH induces transcription of CYP11B2 (coding region) |
Glucocorticoid-suppressive hyperaldosteronism |
| Type 2 | CLCN2 mutations | Increased Cl- efflux activates CYP11B2 transcription |
Early-onset PA |
| Type 3 | KCNJ5 mutations | Increased Na+ influx activates CYP11B2 transcription |
Severe early-onset PA (T158A, I157S, E145Q, G151R) Mild PA (G151E, Y152C) |
| Type 4 | CACNA1H mutations | Increased Ca2+ influx activates CYP11B2 transcription |
Early-onset PA |
9. Other Genes Described in Patients with PA
Somatic mutation of PRKACA, which causes adrenal Cushing’s syndrome, leads to constitutive activation of protein kinase A (PKA), resulting in excess cortisol production [99]. Somatic mutation of PRKACA was reported in a patient with aldosterone and cortisol co-secreting adenoma [100]. Somatic mutation of GNAS, which also causes adrenal Cushing’s syndrome due to constitutive activation of the PKA/cAMP pathway, was reported in two patients with aldosterone and cortisol co-secreting adenoma [101]. Somatic mutations in both genes were also reported in the subsequent study using CYP11B2 IHC-guided targeted NGS, but those mutations were detected in CYP11B2-negative adrenal tumors from APA patients [37][42]. The role of somatic mutation in PRKACA and GNAS in the pathogenesis of PA has not been clarified. Genetic variants of ARMC5, ATP2B4, PDE2A, and PDE2B were indicated to be associated with BHA [102][103][104][105][106].
References
- Funder, J.W.; Carey, R.M.; Mantero, F.; Murad, M.H.; Reincke, M.; Shibata, H.; Stowasser, M.; Young, W.F. The management of primary aldosteronism: Case detection, diagnosis, and treatment: An endocrine society clinical practice guideline. J. Clin. Endocrinol. Metab. 2016, 101, 1889–1916.
- Mulatero, P.; Monticone, S.; Bertello, C.; Viola, A.; Tizzani, D.; Iannaccone, A.; Crudo, V.; Burrello, J.; Milan, A.; Rabbia, F.; et al. Long-Term cardio- and cerebrovascular events in patients with primary aldosteronism. J. Clin. Endocrinol. Metab. 2013, 98, 4826–4833.
- Hundemer, G.L.; Curhan, G.C.; Yozamp, N.; Wang, M.; Vaidya, A. Cardiometabolic outcomes and mortality in medically treated primary aldosteronism: A retrospective cohort study. Lancet Diabetes Endocrinol. 2018, 6, 51–59.
- Hundemer, G.L.; Curhan, G.C.; Yozamp, N.; Wang, M.; Vaidya, A. Renal outcomes in medically and surgically treated primary aldosteronism. Hypertension 2018, 72, 658–666.
- Rossi, G.P.; Maiolino, G.; Flego, A.; Belfiore, A.; Bernini, G.; Fabris, B.; Ferri, C.; Giacchetti, G.; Letizia, C.; Maccario, M.; et al. Adrenalectomy Lowers Incident Atrial Fibrillation in Primary Aldosteronism Patients at Long Term. Hypertension 2018, 71, 585–591.
- Choi, M.; Scholl, U.I.; Yue, P.; Björklund, P.; Zhao, B.; Nelson-Williams, C.; Ji, W.; Cho, Y.; Patel, A.; Men, C.J.; et al. K+ channel mutations in adrenal aldosterone-producing adenomas and hereditary hypertension. Science 2011, 331, 768–772.
- Beuschlein, F.; Boulkroun, S.; Osswald, A.; Wieland, T.; Nielsen, H.N.; Lichtenauer, U.D.; Penton, D.; Schack, V.R.; Amar, L.; Fischer, E.; et al. Somatic mutations in ATP1A1 and ATP2B3 lead to aldosterone-producing adenomas and secondary hypertension. Nat. Genet. 2013, 45, 440–444.
- Azizan, E.A.B.; Poulsen, H.; Tuluc, P.; Zhou, J.; Clausen, M.V.; Lieb, A.; Maniero, C.; Garg, S.; Bochukova, E.G.; Zhao, W.; et al. Somatic mutations in ATP1A1 and CACNA1D underlie a common subtype of adrenal hypertension. Nat. Genet. 2013, 45, 1055–1060.
- Scholl, U.I.; Goh, G.; Stölting, G.; De Oliveira, R.C.; Choi, M.; Overton, J.D.; Fonseca, A.L.; Korah, R.; Starker, L.F.; Kunstman, J.W.; et al. Somatic and germline CACNA1D calcium channel mutations in aldosterone-producing adenomas and primary aldosteronism. Nat. Genet. 2013, 45, 1050–1054.
- Scholl, U.I.; Stölting, G.; Nelson-Williams, C.; Vichot, A.A.; Choi, M.; Loring, E.; Prasad, M.L.; Goh, G.; Carling, T.; Juhlin, C.C.; et al. Recurrent gain of function mutation in calcium channel CACNA1H causes early-onset hypertension with primary aldosteronism. eLife 2015, 4, e06315.
- Scholl, U.I.; Stölting, G.; Schewe, J.; Thiel, A.; Tan, H.; Nelson-Williams, C.; Vichot, A.A.; Jin, S.C.; Loring, E.; Untiet, V.; et al. CLCN2 chloride channel mutations in familial hyperaldosteronism type II. Nat. Genet. 2018, 50, 349–354.
- Fernandes-Rosa, F.L.; Daniil, G.; Orozco, I.J.; Göppner, C.; El Zein, R.; Jain, V.; Boulkroun, S.; Jeunemaitre, X.; Amar, L.; Lefebvre, H.; et al. A gain-of-function mutation in the CLCN2 chloride channel gene causes primary aldosteronism. Nat. Genet. 2018, 50, 355–361.
- Åkerström, T.; Maharjan, R.; Sven Willenberg, H.; Cupisti, K.; Ip, J.; Moser, A.; Stålberg, P.; Robinson, B.; Alexander Iwen, K.; Dralle, H.; et al. Activating mutations in CTNNB1 in aldosterone producing adenomas. Sci. Rep. 2016, 6, 1–9.
- Williams, T.A.; Monticone, S.; Crudo, V.; Warth, R.; Veglio, F.; Mulatero, P. Visinin-like 1 is upregulated in aldosterone-producing adenomas with KCNJ5 mutations and protects from calcium-induced apoptosis. Hypertension 2012, 59, 833–839.
- Kobuke, K.; Oki, K.; Gomez-Sanchez, C.E.; Gomez-Sanchez, E.P.; Ohno, H.; Itcho, K.; Yoshii, Y.; Yoneda, M.; Hattori, N. Calneuron 1 Increased Ca 2+ in the Endoplasmic Reticulum and Aldosterone Production in Aldosterone-Producing Adenoma. Hypertension 2018, 71, 125–133.
- Li, X.; Wang, B.; Tang, L.; Zhang, Y.; Chen, L.; Gu, L.; Zhang, F.; Ouyang, J.; Zhang, X. GSTA1 expression is correlated with aldosterone level in KCNJ5-mutated adrenal aldosterone-producing adenoma. J. Clin. Endocrinol. Metab. 2018, 103, 813–823.
- Der Teo, A.E.; Garg, S.; Johnson, T.I.; Zhao, W.; Zhou, J.; Gomez-Sanchez, C.E.; Gurnell, M.; Brown, M.J. Physiological and Pathological Roles in Human Adrenal of the Glomeruli-Defining Matrix Protein NPNT (Nephronectin). Hypertension 2017, 69, 1207–1216.
- Itcho, K.; Oki, K.; Gomez-Sanchez, C.E.; Gomez-Sanchez, E.P.; Ohno, H.; Kobuke, K.; Nagano, G.; Yoshii, Y.; Baba, R.; Hattori, N.; et al. Endoplasmic Reticulum Chaperone Calmegin Is Upregulated in Aldosterone-Producing Adenoma and Associates with Aldosterone Production. Hypertension 2020, 75, 492–499.
- Howard, B.; Wang, Y.; Xekouki, P.; Faucz, F.R.; Jain, M.; Zhang, L.; Meltzer, P.G.; Stratakis, C.A.; Kebebew, E. Integrated analysis of genome-wide methylation and gene expression shows epigenetic regulation of CYP11B2 in aldosteronomas. J. Clin. Endocrinol. Metab. 2014, 99, 536–543.
- Murakami, M.; Yoshimoto, T.; Nakabayashi, K.; Tsuchiya, K.; Minami, I.; Bouchi, R.; Izumiyama, H.; Fujii, Y.; Abe, K.; Tayama, C.; et al. Integration of transcriptome and methylome analysis of aldosterone-producing adenomas. Eur. J. Endocrinol. 2015, 173, 185–195.
- Yoshii, Y.; Oki, K.; Gomez-Sanchez, C.E.; Ohno, H.; Itcho, K.; Kobuke, K.; Yoneda, M. Hypomethylation of CYP11B2 in Aldosterone-Producing Adenoma. Hypertension 2016, 68, 1432–1437.
- Di Dalmazi, G.; Morandi, L.; Rubin, B.; Pilon, C.; Asioli, S.; Vicennati, V.; De Leo, A.; Ambrosi, F.; Santini, D.; Pagotto, U.; et al. DNA Methylation of Steroidogenic Enzymes in Benign Adrenocortical Tumors: New Insights in Aldosterone-Producing Adenomas. J. Clin. Endocrinol. Metab. 2020, 105, 1–11.
- Sutherland, D.J.; Ruse, J.L.; Laidlaw, J.C. Hypertension, increased aldosterone secretion and low plasma renin activity relieved by dexamethasone. Can. Med. Assoc. J. 1966, 95, 1109–1119.
- Lifton, R.P.; Dluhy, R.G.; Powers, M.; Rich, G.M.; Cook, S.; Ulick, S.; Lalouel, J.-M. A chimaeric llβ-hydroxylase/aldosterone synthase gene causes glucocorticoid-remediable aldosteronism and human hypertension. Nature 1992, 355, 262–265.
- Carroll, J.; Dluhy, R.; Fallo, F.; Pistorello, M.; Bradwin, G.; Gomez-Sanchez, C.E.; Mortensen, R. Aldosterone-producing adenomas do not contain glucocorticoid-remediable aldosteronism chimeric gene duplications. J. Clin. Endocrinol. Metab. 1996, 81, 4310–4312.
- Oki, K.; Plonczynski, M.W.; Lam, M.L.; Gomez-Sanchez, E.P.; Gomez-Sanchez, C.E. Potassium channel mutant KCNJ5 T158A expression in HAC-15 cells increases aldosterone synthesis. Endocrinology 2012, 153, 1774–1782.
- Hattangady, N.G.; Karashima, S.; Yuan, L.; Ponce-Balbuena, D.; Jalife, J.; Gomez-Sanchez, C.E.; Auchus, R.J.; Rainey, W.E.; Else, T. Mutated KCNJ5 activates the acute and chronic regulatory steps in aldosterone production. J. Mol. Endocrinol. 2016, 57, 1–11.
- Tauber, P.; Penton, D.; Stindl, J.; Humberg, E.; Tegtmeier, I.; Sterner, C.; Beuschlein, F.; Reincke, M.; Barhanin, J.; Bandulik, S.; et al. Pharmacology and pathophysiology of mutated KCNJ5 found in adrenal aldosterone-producing adenomas. Endocrinology 2014, 155, 1353–1362.
- Kuppusamy, M.; Caroccia, B.; Stindl, J.; Bandulik, S.; Lenzini, L.; Gioco, F.; Fishman, V.; Zanotti, G.; Gomez-Sanchez, C.; Bader, M.; et al. A novel KCNJ5-insT149 somatic mutation close to, but outside, the selectivity filter causes resistant hypertension by loss of selectivity for potassium. J. Clin. Endocrinol. Metab. 2014, 99, E1765–E1773.
- Monticone, S.; Hattangady, N.G.; Nishimoto, K.; Mantero, F.; Rubin, B.; Cicala, M.V.; Pezzani, R.; Auchus, R.J.; Ghayee, H.K.; Shibata, H.; et al. Effect of KCNJ5 mutations on gene expression in aldosterone-producing adenomas and adrenocortical cells. J. Clin. Endocrinol. Metab. 2012, 97, 1567–1572.
- Oki, K.; Gomez-Sanchez, C.E. The landscape of molecular mechanism for aldosterone production in aldosterone-producing adenoma. Endocr. J. 2020, 67, 989–995.
- Boulkroun, S.; Golib Dzib, J.F.; Samson-Couterie, B.; Rosa, F.L.F.; Rickard, A.J.; Meatchi, T.; Amar, L.; Benecke, A.; Zennaro, M.C. KCNJ5 mutations in aldosterone producing adenoma and relationship with adrenal cortex remodeling. Mol. Cell. Endocrinol. 2013, 371, 221–227.
- Yang, Y.; Gomez-Sanchez, C.E.; Jaquin, D.; Aristizabal Prada, E.T.; Meyer, L.S.; Knösel, T.; Schneider, H.; Beuschlein, F.; Reincke, M.; Williams, T.A. Primary Aldosteronism: KCNJ5 Mutations and Adrenocortical Cell Growth. Hypertension 2019, 74, 809–816.
- Cheng, C.J.; Sung, C.C.; Wu, S.T.; Lin, Y.C.; Sytwu, H.K.; Huang, C.L.; Lin, S.H. Novel KCNJ5 mutations in sporadic aldosterone-producing adenoma reduce Kir3.4 membrane abundance. J. Clin. Endocrinol. Metab. 2015, 100, E155–E163.
- Williams, T.A.; Monticone, S.; Schack, V.R.; Stindl, J.; Burrello, J.; Buffolo, F.; Annaratone, L.; Castellano, I.; Beuschlein, F.; Reincke, M.; et al. Somatic ATP1A1, ATP2B3, and KCNJ5 mutations in aldosterone-producing adenomas. Hypertension 2014, 63, 188–195.
- Hardege, I.; Xu, S.; Gordon, R.D.; Thompson, A.J.; Figg, N.; Stowasser, M.; Murrell-Lagnado, R.; O’Shaughnessy, K.M. Novel insertion mutation in KCNJ5 channel produces constitutive aldosterone release from H295R cells. Mol. Endocrinol. 2015, 29, 1522–1530.
- Nanba, K.; Omata, K.; Else, T.; Beck, P.C.C.; Nanba, A.T.; Turcu, A.F.; Miller, B.S.; Giordano, T.J.; Tomlins, S.A.; Rainey, W.E. Targeted molecular characterization of aldosterone-producing adenomas in white americans. J. Clin. Endocrinol. Metab. 2018, 103, 3869–3876.
- Scholl, U.I.; Healy, J.M.; Thiel, A.; Fonseca, A.L.; Brown, T.C.; Kunstman, J.W.; Horne, M.J.; Dietrich, D.; Riemer, J.; Kücükköylü, S.; et al. Novel somatic mutations in primary hyperaldosteronism are related to the clinical, radiological and pathological phenotype. Clin. Endocrinol. 2015, 83, 779–789.
- Åkerström, T.; Crona, J.; Delgado Verdugo, A.; Starker, L.F.; Cupisti, K.; Willenberg, H.S.; Knoefel, W.T.; Saeger, W.; Feller, A.; Ip, J.; et al. Comprehensive re-sequencing of adrenal aldosterone producing lesions reveal three somatic mutations near the KCNJ5 potassium channel selectivity filter. PLoS ONE 2012, 7, e41926.
- Nanba, K.; Omata, K.; Tomlins, S.A.; Giordano, T.J.; Hammer, G.D.; Rainey, W.E.; Else, T. Double adrenocortical adenomas harboring independent KCNJ5 and PRKACA somatic mutations. Eur. J. Endocrinol. 2016, 175, K1–K6.
- Fang-Fang, Z.; Li-Min, Z.; Ai-Fang, N.; Xiao-Ying, L.; Jing-Rong, L.; Ke, Z.; Jing, C.; Wen-Long, Z.; Zhou-Jun, S.; Yi-Chun, Z.; et al. Clinical Characteristics of Somatic Mutations in Chinese Patients With Aldosterone-Producing Adenoma. Hypertension 2015, 65, 622–628.
- Nanba, K.; Omata, K.; Gomez-Sanchez, C.E.; Stratakis, C.A.; Demidowich, A.P.; Suzuki, M.; Thompson, L.D.R.; Cohen, D.L.; Luther, J.M.; Gellert, L.; et al. Genetic characteristics of aldosterone-producing adenomas in blacks. Hypertension 2019, 73, 885–892.
- Kitamoto, T.; Omura, M.; Suematsu, S.; Saito, J.; Nishikawa, T. KCNJ5 mutation as a predictor for resolution of hypertension after surgical treatment of aldosterone-producing adenoma. J. Hypertens. 2018, 36, 619–627.
- Azizan, E.A.B.; Murthy, M.; Stowasser, M.; Gordon, R.; Kowalski, B.; Xu, S.; Brown, M.J.; O’Shaughnessy, K.M. Somatic mutations affecting the selectivity filter of KCNJ5 are frequent in 2 large unselected collections of adrenal aldosteronomas. Hypertension 2012, 59, 587–591.
- Fernandes-Rosa, F.L.; Williams, T.A.; Riester, A.; Steichen, O.; Beuschlein, F.; Boulkroun, S.; Strom, T.M.; Monticone, S.; Amar, L.; Meatchi, T.; et al. Genetic spectrum and clinical correlates of somatic mutations in aldosterone-producing adenoma. Hypertension 2014, 64, 354–361.
- Taguchi, R.; Yamada, M.; Nakajima, Y.; Satoh, T.; Hashimoto, K.; Shibusawa, N.; Ozawa, A.; Okada, S.; Rokutanda, N.; Takata, D.; et al. Expression and mutations of KCNJ5 mRNA in Japanese patients with aldosterone-producing adenomas. J. Clin. Endocrinol. Metab. 2012, 97, 1311–1319.
- Ram Hong, A.; Kim, J.H.; Song, Y.S.; Lee, K.E.; Seo, S.H.; Seong, M.W.; Shin, C.S.; Kim, S.W.; Kim, S.Y. Genetics of aldosterone-producing adenoma in Korean patients. PLoS ONE 2016, 11, e0147590.
- Okamura, T.; Nakajima, Y.; Katano-Toki, A.; Horiguchi, K.; Matsumoto, S.; Yoshino, S.; Yamada, E.; Tomaru, T.; Ishii, S.; Saito, T.; et al. Characteristics of japanese aldosterone-producing adenomas with KCNJ5 mutations. Endocr. J. 2017, 64, 39–47.
- Warachit, W.; Atikankul, T.; Houngngam, N.; Sunthornyothin, S. Prevalence of somatic KCNJ5 mutations in Thai patients with aldosterone-producing adrenal adenomas. J. Endocr. Soc. 2018, 2, 1137–1146.
- Wu, V.C.; Huang, K.H.; Peng, K.Y.; Tsai, Y.C.; Wu, C.H.; Wang, S.M.; Yang, S.Y.; Lin, L.Y.; Chang, C.C.; Lin, Y.H.; et al. Prevalence and clinical correlates of somatic mutation in aldosterone producing adenoma-Taiwanese population. Sci. Rep. 2015, 5, 1–10.
- Lenzini, L.; Rossitto, G.; Maiolino, G.; Letizia, C.; Funder, J.W.; Rossi, G.P. A meta-analysis of somatic KCNJ5 K+ channel mutations in 1636 patients with an aldosterone-producing adenoma. J. Clin. Endocrinol. Metab. 2015, 100, E1089–E1095.
- Yamada, M.; Nakajima, Y.; Taguchi, R.; Okamura, T.; Ishii, S.; Tomaru, T.; Ozawa, A.; Shibusawa, N.; Yoshino, S.; Toki, A.; et al. KCNJ5 mutations in aldosterone- and cortisol-co-secreting adrenal adenomas. Endocr. J. 2012, 59, 735–741.
- Peng, K.Y.; Liao, H.W.; Chan, C.K.; Lin, W.C.; Yang, S.Y.; Tsai, Y.C.; Huang, K.H.; Lin, Y.H.; Chueh, J.S.; Wu, V.C. Presence of subclinical hypercortisolism in clinical aldosterone-producing adenomas predicts lower clinical success. Hypertension 2020, 76, 1537–1544.
- Rossi, G.P.; Cesari, M.; Letizia, C.; Seccia, T.M.; Cicala, M.V.; Zinnamosca, L.; Kuppusamy, M.; Mareso, S.; Sciomer, S.; Iacobone, M.; et al. KCNJ5 gene somatic mutations affect cardiac remodelling but do not preclude cure of high blood pressure and regression of left ventricular hypertrophy in primary aldosteronism. J. Hypertens. 2014, 32, 1514–1522.
- Kitamoto, T.; Suematsu, S.; Matsuzawa, Y.; Saito, J.; Omura, M.; Nishikawa, T. Comparison of cardiovascular complications in patients with and without KCNJ5 gene mutations harboring aldosterone-producing adenomas. J. Atheroscler. Thromb. 2015, 22, 191–200.
- Chang, Y.-Y.; Tsai, C.-H.; Peng, S.-Y.; Chen, Z.-W.; Chang, C.-C.; Lee, B.-C.; Liao, C.-W.; Pan, C.-T.; Chen, Y.-L.; Lin, L.-C.; et al. KCNJ5 Somatic Mutations in Aldosterone-Producing Adenoma Are Associated With a Worse Baseline Status and Better Recovery of Left Ventricular Remodeling and Diastolic Function. Hypertension 2021, 77, 114–125.
- Vilela, L.A.P.; Rassi-Cruz, M.; Guimaraes, A.G.; Moises, C.C.S.; Freitas, T.C.; Alencar, N.P.; Petenuci, J.; Goldbaum, T.S.; MacIel, A.A.W.; Pereira, M.A.A.; et al. KCNJ5 Somatic Mutation Is a Predictor of Hypertension Remission after Adrenalectomy for Unilateral Primary Aldosteronism. J. Clin. Endocrinol. Metab. 2019, 104, 4695–4702.
- Nanba, K.; Chen, A.X.; Omata, K.; Vinco, M.; Giordano, T.J.; Else, T.; Hammer, G.D.; Tomlins, S.A.; Rainey, W.E. Molecular heterogeneity in aldosterone-producing adenomas. J. Clin. Endocrinol. Metab. 2016, 101, 999–1007.
- Azizan, E.A.B.; Lam, B.Y.H.; Newhouse, S.J.; Zhou, J.; Kuc, R.E.; Clarke, J.; Happerfield, L.; Marker, A.; Hoffman, G.J.; Brown, M.J. Microarray, qPCR, and KCNJ5 sequencing of aldosterone-producing adenomas reveal differences in genotype and phenotype between zona glomerulosa- and zona fasciculata-like tumors. J. Clin. Endocrinol. Metab. 2012, 97, 819–829.
- Inoue, K.; Yamazaki, Y.; Kitamoto, T.; Hirose, R.; Saito, J.; Omura, M.; Sasano, H.; Nishikawa, T. Aldosterone Suppression by Dexamethasone in Patients with KCNJ5-Mutated Aldosterone-Producing Adenoma. J. Clin. Endocrinol. Metab. 2018, 103, 3477–3485.
- Ono, Y.; Yamazaki, Y.; Omata, K.; Else, T.; Tomlins, S.A.; Rhayem, Y.; Williams, T.A.; Reincke, M.; Carling, T.; Monticone, S.; et al. Histological Characterization of Aldosterone-producing Adrenocortical Adenomas with Different Somatic Mutations. J. Clin. Endocrinol. Metab. 2020, 105, e282–e289.
- Williams, T.A.; Peitzsch, M.; Dietz, A.S.; Dekkers, T.; Bidlingmaier, M.; Riester, A.; Treitl, M.; Rhayem, Y.; Beuschlein, F.; Lenders, J.W.M.; et al. Genotype-Specific Steroid Profiles Associated With Aldosterone-Producing Adenomas. Hypertension 2016, 67, 139–145.
- Tezuka, Y.; Yamazaki, Y.; Kitada, M.; Morimoto, R.; Kudo, M.; Seiji, K.; Takase, K.; Kawasaki, Y.; Mitsuzuka, K.; Ito, A.; et al. 18-Oxocortisol Synthesis in Aldosterone-Producing Adrenocortical Adenoma and Significance of KCNJ5 Mutation Status. Hypertension 2019, 73, 1283–1290.
- Satoh, F.; Morimoto, R.; Ono, Y.; Iwakura, Y.; Omata, K.; Kudo, M.; Takase, K.; Seiji, K.; Sasamoto, H.; Honma, S.; et al. Measurement of peripheral plasma 18-oxocortisol can discriminate unilateral adenoma from bilateral diseases in patients with primary aldosteronism. Hypertension 2015, 65, 1096–1102.
- Eisenhofer, G.; Dekkers, T.; Peitzsch, M.; Dietz, A.S.; Bidlingmaier, M.; Treitl, M.; Williams, T.A.; Bornstein, S.R.; Haase, M.; Rump, L.C.; et al. Mass spectrometry-based adrenal and peripheral venous steroid profiling for subtyping primary aldosteronism. Clin. Chem. 2016, 62, 514–524.
- Turcu, A.F.; Wannachalee, T.; Tsodikov, A.; Nanba, A.T.; Ren, J.; Shields, J.J.; O’Day, P.J.; Giacherio, D.; Rainey, W.E.; Auchus, R.J. Comprehensive Analysis of Steroid Biomarkers for Guiding Primary Aldosteronism Subtyping. Hypertension 2020, 75, 183–192.
- Eisenhofer, G.; Durán, C.; Cannistraci, C.V.; Peitzsch, M.; Williams, T.A.; Riester, A.; Burrello, J.; Buffolo, F.; Prejbisz, A.; Beuschlein, F.; et al. Use of Steroid Profiling Combined With Machine Learning for Identification and Subtype Classification in Primary Aldosteronism. JAMA Netw. Open 2020, 3, e2016209.
- Geller, D.S.; Zhang, J.; Wisgerhof, M.V.; Shackleton, C.; Kashgarian, M.; Lifton, R.P. A novel form of human mendelian hypertension featuring nonglucocorticoid- remediable aldosteronism. J. Clin. Endocrinol. Metab. 2008, 93, 3117–3123.
- Charmandari, E.; Sertedaki, A.; Kino, T.; Merakou, C.; Hoffman, D.A.; Hatch, M.M.; Hurt, D.E.; Lin, L.; Xekouki, P.; Stratakis, C.A.; et al. A novel point mutation in the KCNJ5 gene causing primary hyperaldosteronism and early-onset autosomal dominant hypertension. J. Clin. Endocrinol. Metab. 2012, 97, 1532–1539.
- Monticone, S.; Bandulik, S.; Stindl, J.; Zilbermint, M.; Dedov, I.; Mulatero, P.; Allgaeuer, M.; Lee, C.C.R.; Stratakis, C.A.; Williams, T.A.; et al. A case of severe hyperaldosteronism caused by a de novo mutation affecting a critical salt bridge Kir3.4 residue. J. Clin. Endocrinol. Metab. 2015, 100, E114–E118.
- Scholl, U.I.; Nelson-Williams, C.; Yue, P.; Grekin, R.; Wyatt, R.J.; Dillon, M.J.; Couch, R.; Hammer, L.K.; Harley, F.L.; Farhi, A.; et al. Hypertension with or without adrenal hyperplasia due to different inherited mutations in the potassium channel KCNJ5. Proc. Natl. Acad. Sci. USA 2012, 109, 2533–2538.
- Mulatero, P.; Tauber, P.; Zennaro, M.C.; Monticone, S.; Lang, K.; Beuschlein, F.; Fischer, E.; Tizzani, D.; Pallauf, A.; Viola, A.; et al. KCNJ5 mutations in European families with nonglucocorticoid remediable familial hyperaldosteronism. Hypertension 2012, 59, 235–240.
- Monticone, S.; Hattangady, N.G.; Penton, D.; Isales, C.M.; Edwards, M.A.; Williams, T.A.; Sterner, C.; Warth, R.; Mulatero, P.; Rainey, W.E. A novel Y152C KCNJ5 mutation responsible for familial hyperaldosteronism type III. J. Clin. Endocrinol. Metab. 2013, 98, 1861–1865.
- Adachi, M.; Muroya, K.; Asakura, Y.; Sugiyama, K.; Homma, K.; Hasegawa, T. Discordant genotype-phenotype correlation in familial hyperaldosteronism type III with KCNJ5 gene mutation: A patient report and review of the literature. Horm. Res. Paediatr. 2014, 82, 138–142.
- Tamura, A.; Nishimoto, K.; Seki, T.; Matsuzawa, Y.; Saito, J.; Omura, M.; Gomez-Sanchez, C.E.; Makita, K.; Matsui, S.; Moriya, N.; et al. Somatic KCNJ5 mutation occurring early in adrenal development may cause a novel form of juvenile primary aldosteronism. Mol. Cell. Endocrinol. 2017, 441, 134–139.
- Maria, A.G.; Suzuki, M.; Berthon, A.; Kamilaris, C.; Demidowich, A.; Lack, J.; Zilbermint, M.; Hannah-Shmouni, F.; Faucz, F.R.; Stratakis, C.A. Mosaicism for KCNJ5 Causing Early-Onset Primary Aldosteronism due to Bilateral Adrenocortical Hyperplasia. Am. J. Hypertens. 2020, 33, 124–130.
- Stindl, J.; Tauber, P.; Sterner, C.; Tegtmeier, I.; Warth, R.; Bandulik, S. Pathogenesis of adrenal aldosterone-producing adenomas carrying mutations of the Na+/K+-ATPase. Endocrinology 2015, 156, 4582–4591.
- Nanba, K.; Yamazaki, Y.; Bick, N.; Onodera, K.; Tezuka, Y.; Omata, K.; Ono, Y.; Blinder, A.R.; Tomlins, S.A.; Rainey, W.E.; et al. Prevalence of Somatic Mutations in Aldosterone-Producing Adenomas in Japanese Patients. J. Clin. Endocrinol. Metab. 2020, 105, 1–8.
- De Sousa, K.; Boulkroun, S.; Baron, S.; Nanba, K.; Wack, M.; Rainey, W.E.; Rocha, A.; Giscos-Douriez, I.; Meatchi, T.; Amar, L.; et al. Genetic, cellular, and molecular heterogeneity in adrenals with aldosterone-producing adenoma. Hypertension 2020, 75, 1034–1044.
- Kitamoto, T.; Suematsu, S.; Yamazaki, Y.; Nakamura, Y.; Sasano, H.; Matsuzawa, Y.; Saito, J.; Omura, M.; Nishikawa, T. Clinical and steroidogenic characteristics of aldosterone-producing adenomas with ATPase or CACNA1D gene mutations. J. Clin. Endocrinol. Metab. 2016, 101, 494–503.
- Backman, S.; Åkerström, T.; Maharjan, R.; Cupisti, K.; Willenberg, H.S.; Hellman, P.; Björklund, P. RNA Sequencing Provides Novel Insights into the Transcriptome of Aldosterone Producing Adenomas. Sci. Rep. 2019, 9, 1–10.
- Tauber, P.; Aichinger, B.; Christ, C.; Stindl, J.; Rhayem, Y.; Beuschlein, F.; Warth, R.; Bandulik, S. Cellular pathophysiology of an adrenal adenoma-associated mutant of the plasma membrane Ca2+-ATPase ATP2B3. Endocrinology 2016, 157, 2489–2499.
- Dekkers, T.; Ter Meer, M.; Lenders, J.W.M.; Hermus, A.R.M.; Schultze Kool, L.; Langenhuijsen, J.F.; Nishimoto, K.; Ogishima, T.; Mukai, K.; Azizan, E.A.B.; et al. Adrenal nodularity and somatic mutations in primary aldosteronism: One node is the culprit? J. Clin. Endocrinol. Metab. 2014, 99, 1341–1351.
- Ortner, N.J.; Kaserer, T.; Copeland, J.N.; Striessnig, J. De novo CACAN1D Ca2+ channelopathies: Clinical phenotypes and molecular mechanism. Pflug. Arch. Eur. J. Physiol. 2020, 472, 755–773.
- Semenova, N.A.; Ryzhkova, O.R.; Strokova, T.V.; Taran, N.N. The third case report a patient with primary aldosteronism, seizures, and neurologic abnormalities (PASNA) syndrome de novo variant mutations in the CACNA1D gene. Zhurnal Nevrol. Psikhiatrii Im. S.S. Korsakova 2018, 118, 49–52.
- De Mingo Alemany, M.C.; Mifsud Grau, L.; Moreno Macián, F.; Ferrer Lorente, B.; León Cariñena, S. A de novo CACNA1D missense mutation in a patient with congenital hyperinsulinism, primary hyperaldosteronism and hypotonia. Channels 2020, 14, 175–180.
- Kim, A.C.; Reuter, A.L.; Zubair, M.; Else, T.; Serecky, K.; Bingham, N.C.; Lavery, G.G.; Parker, K.L.; Hammer, G.D. Targeted disruption β-catenin in Sf1-expressing cells impairs development and maintenance of the adrenal cortex. Development 2008, 135, 2593–2602.
- Boulkroun, S.; Samson-Couterie, B.; Golib-Dzib, J.F.; Amar, L.; Plouin, P.F.; Sibony, M.; Lefebvre, H.; Louiset, E.; Jeunemaitre, X.; Meatchi, T.; et al. Aldosterone-producing adenoma formation in the adrenal cortex involves expression of stem/progenitor cell markers. Endocrinology 2011, 152, 4753–4763.
- Berthon, A.; Drelon, C.; Ragazzon, B.; Boulkroun, S.; Tissier, F.; Amar, L.; Samson-Couterie, B.; Zennaro, M.C.; Plouin, P.F.; Skah, S.; et al. WNT/β-catenin signalling is activated in aldosteroneproducing adenomas and controls aldosterone production. Hum. Mol. Genet. 2014, 23, 889–905.
- Wu, V.C.; Wang, S.M.; Chueh, S.C.J.; Yang, S.Y.; Huang, K.H.; Lin, Y.H.; Wang, J.J.; Connolly, R.; Hu, Y.H.; Gomez-Sanchez, C.E.; et al. The prevalence of CTNNB1 mutations in primary aldosteronism and consequences for clinical outcomes. Sci. Rep. 2017, 7, 1–10.
- Tissier, F.; Cavard, C.; Groussin, L.; Perlemoine, K.; Fumey, G.; Hagneré, A.M.; René-Corail, F.; Jullian, E.; Gicquel, C.; Bertagna, X.; et al. Mutations of β-catenin in adrenocortical tumors: Activation of the Wnt signaling pathway is a frequent event in both benign and malignant adrenocortical tumors. Cancer Res. 2005, 65, 7622–7627.
- Berthon, A.; Sahut-Barnola, I.; Lambert-Langlais, S.; de Joussineau, C.; Damon-Soubeyrand, C.; Louiset, E.; Taketo, M.M.; Tissier, F.; Bertherat, J.; Lefrançois-Martinez, A.M.; et al. Constitutive β-catenin activation induces adrenal hyperplasia and promotes adrenal cancer development. Hum. Mol. Genet. 2010, 19, 1561–1576.
- Gordon, R.D.; Stowasser, M.; Tunny, T.J.; Klemm, S.A.; Finn, W.L.; Krek, A.L. Clinical and pathological diversity of primary aldosteronism, including a new familial variety. Clin. Exp. Pharmacol. Physiol. 1991, 18, 283–286.
- Stowasser, M.; Gordon, R.D.; Tunny, T.J.; Klemm, S.A.; Finn, W.L.; Krek, A.L. Familial hyperaldosteronism type II: Five families with a new variety of primary aldosteronism. Clin. Exp. Pharmacol. Physiol. 1992, 19, 319–322.
- Dutta, R.K.; Arnesen, T.; Heie, A.; Walz, M.; Alesina, P.; Söderkvist, P.; Gimm, O. A somatic mutation in CLCN2 identified in a sporadic aldosterone-producing adenoma. Eur. J. Endocrinol. 2019, 181, K37–K41.
- Rege, J.; Nanba, K.; Blinder, A.R.; Plaska, S.; Udager, A.M.; Vats, P.; Kumar-Sinha, C.; Giordano, T.J.; Rainey, W.E.; Else, T. Identification of Somatic Mutations in CLCN2 in Aldosterone-Producing Adenomas. J. Endocr. Soc. 2020, 4, bvaa123.
- Daniil, G.; Fernandes-Rosa, F.L.; Chemin, J.; Blesneac, I.; Beltrand, J.; Polak, M.; Jeunemaitre, X.; Boulkroun, S.; Amar, L.; Strom, T.M.; et al. CACNA1H Mutations Are Associated With Different Forms of Primary Aldosteronism. EBioMedicine 2016, 13, 225–236.
- Nanba, K.; Blinder, A.R.; Rege, J.; Hattangady, N.G.; Else, T.; Liu, C.J.; Tomlins, S.A.; Vats, P.; Kumar-Sinha, C.; Giordano, T.J.; et al. Somatic CACNA1H Mutation As a Cause of Aldosterone-Producing Adenoma. Hypertension 2020, 75, 645–649.
- Beuschlein, F.; Fassnacht, M.; Assié, G.; Calebiro, D.; Stratakis, C.A.; Osswald, A.; Ronchi, C.L.; Wieland, T.; Sbiera, S.; Faucz, F.R.; et al. Constitutive Activation of PKA Catalytic Subunit in Adrenal Cushing’s Syndrome. N. Engl. J. Med. 2014, 370, 1019–1028.
- Rhayem, Y.; Perez-Rivas, L.G.; Dietz, A.; Bathon, K.; Gebhard, C.; Riester, A.; Mauracher, B.; Gomez-Sanchez, C.; Eisenhofer, G.; Schwarzmayr, T.; et al. PRKACA somatic mutations are rare findings in aldosterone-producing adenomas. J. Clin. Endocrinol. Metab. 2016, 101, 3010–3017.
- Nakajima, Y.; Okamura, T.; Horiguchi, K.; Gohko, T.; Miyamoto, T.; Satoh, T.; Ozawa, A.; Ishii, S.; Yamada, E.; Hashimoto, K.; et al. Gnas mutations in adrenal aldosterone-producing adenomas. Endocr. J. 2016, 63, 199–204.
- Zilbermint, M.; Xekouki, P.; Faucz, F.R.; Berthon, A.; Gkourogianni, A.; Schernthaner-Reiter, M.H.; Batsis, M.; Sinaii, N.; Quezado, M.M.; Merino, M.; et al. Primary aldosteronism and ARMC5 variants. J. Clin. Endocrinol. Metab. 2015, 100, E900–E909.
- Mulatero, P.; Schiavi, F.; Williams, T.A.; Monticone, S.; Barbon, G.; Opocher, G.; Fallo, F. ARMC5 mutation analysis in patients with primary aldosteronism and bilateral adrenal lesions. J. Hum. Hypertens. 2016, 30, 374–378.
- Joseph, J.J.; Zhou, X.; Zilbermint, M.; Stratakis, C.A.; Faucz, F.R.; Lodish, M.B.; Berthon, A.; Wilson, J.G.; Hsueh, W.A.; Golden, S.H.; et al. The Association of ARMC5 with the Renin-Angiotensin-Aldosterone System, Blood Pressure, and Glycemia in African Americans. J. Clin. Endocrinol. Metab. 2020, 105, 2625–2633.
- Hattangady, N.G.; Foster, J.; Lerario, A.M.; Ponce-Balbuena, D.; Rege, J.; Monticone, S.; Rainey, W.E.; Mulatero, P.; Else, T. Molecular and Electrophysiological Analyses of ATP2B4 Gene Variants in Bilateral Adrenal Hyperaldosteronism. Horm. Cancer 2020, 11, 52–62.
- Rassi-Cruz, M.; Maria, A.G.; Faucz, F.R.; London, E.; Vilela, L.A.P.; Santana, L.S.; Benedetti, A.F.F.; Goldbaum, T.S.; Tanno, F.Y.; Srougi, V.; et al. Phosphodiesterase 2A and 3B variants are associated with primary aldosteronism. Endocr. Relat. Cancer 2021, 28, 1–13.


