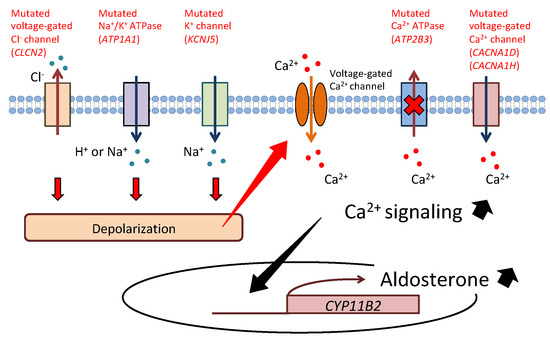
Primary aldosteronism (PA) is the most common form of secondary hypertension, with a prevalence of 5–10% among patients with hypertension. PA is mainly classified into two subtypes: aldosterone-producing adenoma (APA) and bilateral idiopathic hyperaldosteronism. Recent developments in genetic analysis have facilitated the discovery of mutations in KCNJ5, ATP1A1, ATP2B3, CACNA1D, CACNA1H, CLCN2, and CTNNB1 in sporadic or familial forms of PA in the last decade. These findings have greatly advanced our understanding of the mechanism of excess aldosterone synthesis, particularly in APA. Most of the causative genes encode ion channels or pumps, and their mutations lead to depolarization of the cell membrane due to impairment of ion transport. Depolarization activates voltage-gated Ca2+ channels and intracellular calcium signaling and promotes the transcription of aldosterone synthase, resulting in overproduction of aldosterone.
- primary aldosteronism
- hypertension
- somatic mutation
- aldosterone-producing adenoma
1. Primary aldosteronism
2. KCNJ5
3. ATP1A1
4. ATP2B3
5. CACNA1D
6. CTNNB1
| Gene | Clinical Characteristics | Histological Features |
|---|---|---|
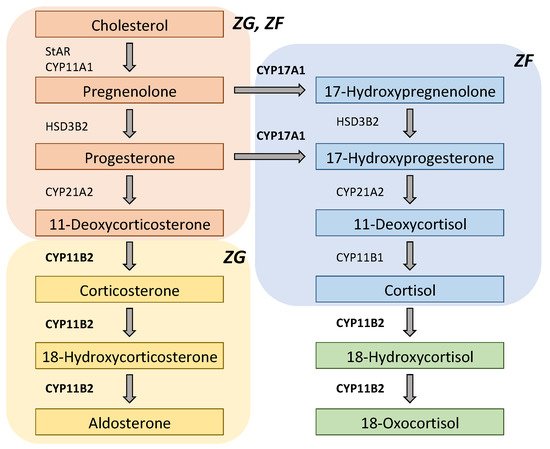
| KCNJ5 | More common in Asians More often female Diagnosed at younger age Larger tumor size Higher plasma levels of aldosterone, 18-oxocortisol, and 18-hydroxycortisol More likely to have hypertension remission after adrenalectomy |
Clear cell dominant (ZF-like) |
| ATP1A1 | More often male Smaller tumor size |
Compact cell dominant (ZG-like) |
| ATP2B3 | More often male Smaller tumor size |
Compact cell dominant (ZG-like) |
| CACNA1D | More common in African Americans More often male Smaller tumor size |
Compact cell dominant (ZG-like) |
| CTNNB1 | More often female Higher risk of post adrenalectomy residual hypertension |
Variable |
7. CLCN2
8. CACNA1H
| Genetic Variant | Molecular Mechanism | Clinical Characteristics | |
|---|---|---|---|
| Type 1 | CYP11B1/CYP11B2 chimeric gene |
ACTH induces transcription of CYP11B2 (coding region) |
Glucocorticoid-suppressive hyperaldosteronism |
| Type 2 | CLCN2 mutations | Increased Cl- efflux activates CYP11B2 transcription |
Early-onset PA |
| Type 3 | KCNJ5 mutations | Increased Na+ influx activates CYP11B2 transcription |
Severe early-onset PA (T158A, I157S, E145Q, G151R) Mild PA (G151E, Y152C) |
| Type 4 | CACNA1H mutations | Increased Ca2+ influx activates CYP11B2 transcription |
Early-onset PA |
9. Other Genes Described in Patients with PA
This entry is adapted from the peer-reviewed paper 10.3390/biomedicines9040409
References
- Funder, J.W.; Carey, R.M.; Mantero, F.; Murad, M.H.; Reincke, M.; Shibata, H.; Stowasser, M.; Young, W.F. The management of primary aldosteronism: Case detection, diagnosis, and treatment: An endocrine society clinical practice guideline. J. Clin. Endocrinol. Metab. 2016, 101, 1889–1916.
- Mulatero, P.; Monticone, S.; Bertello, C.; Viola, A.; Tizzani, D.; Iannaccone, A.; Crudo, V.; Burrello, J.; Milan, A.; Rabbia, F.; et al. Long-Term cardio- and cerebrovascular events in patients with primary aldosteronism. J. Clin. Endocrinol. Metab. 2013, 98, 4826–4833.
- Hundemer, G.L.; Curhan, G.C.; Yozamp, N.; Wang, M.; Vaidya, A. Cardiometabolic outcomes and mortality in medically treated primary aldosteronism: A retrospective cohort study. Lancet Diabetes Endocrinol. 2018, 6, 51–59.
- Hundemer, G.L.; Curhan, G.C.; Yozamp, N.; Wang, M.; Vaidya, A. Renal outcomes in medically and surgically treated primary aldosteronism. Hypertension 2018, 72, 658–666.
- Rossi, G.P.; Maiolino, G.; Flego, A.; Belfiore, A.; Bernini, G.; Fabris, B.; Ferri, C.; Giacchetti, G.; Letizia, C.; Maccario, M.; et al. Adrenalectomy Lowers Incident Atrial Fibrillation in Primary Aldosteronism Patients at Long Term. Hypertension 2018, 71, 585–591.
- Choi, M.; Scholl, U.I.; Yue, P.; Björklund, P.; Zhao, B.; Nelson-Williams, C.; Ji, W.; Cho, Y.; Patel, A.; Men, C.J.; et al. K+ channel mutations in adrenal aldosterone-producing adenomas and hereditary hypertension. Science 2011, 331, 768–772.
- Beuschlein, F.; Boulkroun, S.; Osswald, A.; Wieland, T.; Nielsen, H.N.; Lichtenauer, U.D.; Penton, D.; Schack, V.R.; Amar, L.; Fischer, E.; et al. Somatic mutations in ATP1A1 and ATP2B3 lead to aldosterone-producing adenomas and secondary hypertension. Nat. Genet. 2013, 45, 440–444.
- Azizan, E.A.B.; Poulsen, H.; Tuluc, P.; Zhou, J.; Clausen, M.V.; Lieb, A.; Maniero, C.; Garg, S.; Bochukova, E.G.; Zhao, W.; et al. Somatic mutations in ATP1A1 and CACNA1D underlie a common subtype of adrenal hypertension. Nat. Genet. 2013, 45, 1055–1060.
- Scholl, U.I.; Goh, G.; Stölting, G.; De Oliveira, R.C.; Choi, M.; Overton, J.D.; Fonseca, A.L.; Korah, R.; Starker, L.F.; Kunstman, J.W.; et al. Somatic and germline CACNA1D calcium channel mutations in aldosterone-producing adenomas and primary aldosteronism. Nat. Genet. 2013, 45, 1050–1054.
- Scholl, U.I.; Stölting, G.; Nelson-Williams, C.; Vichot, A.A.; Choi, M.; Loring, E.; Prasad, M.L.; Goh, G.; Carling, T.; Juhlin, C.C.; et al. Recurrent gain of function mutation in calcium channel CACNA1H causes early-onset hypertension with primary aldosteronism. eLife 2015, 4, e06315.
- Scholl, U.I.; Stölting, G.; Schewe, J.; Thiel, A.; Tan, H.; Nelson-Williams, C.; Vichot, A.A.; Jin, S.C.; Loring, E.; Untiet, V.; et al. CLCN2 chloride channel mutations in familial hyperaldosteronism type II. Nat. Genet. 2018, 50, 349–354.
- Fernandes-Rosa, F.L.; Daniil, G.; Orozco, I.J.; Göppner, C.; El Zein, R.; Jain, V.; Boulkroun, S.; Jeunemaitre, X.; Amar, L.; Lefebvre, H.; et al. A gain-of-function mutation in the CLCN2 chloride channel gene causes primary aldosteronism. Nat. Genet. 2018, 50, 355–361.
- Åkerström, T.; Maharjan, R.; Sven Willenberg, H.; Cupisti, K.; Ip, J.; Moser, A.; Stålberg, P.; Robinson, B.; Alexander Iwen, K.; Dralle, H.; et al. Activating mutations in CTNNB1 in aldosterone producing adenomas. Sci. Rep. 2016, 6, 1–9.
- Williams, T.A.; Monticone, S.; Crudo, V.; Warth, R.; Veglio, F.; Mulatero, P. Visinin-like 1 is upregulated in aldosterone-producing adenomas with KCNJ5 mutations and protects from calcium-induced apoptosis. Hypertension 2012, 59, 833–839.
- Kobuke, K.; Oki, K.; Gomez-Sanchez, C.E.; Gomez-Sanchez, E.P.; Ohno, H.; Itcho, K.; Yoshii, Y.; Yoneda, M.; Hattori, N. Calneuron 1 Increased Ca 2+ in the Endoplasmic Reticulum and Aldosterone Production in Aldosterone-Producing Adenoma. Hypertension 2018, 71, 125–133.
- Li, X.; Wang, B.; Tang, L.; Zhang, Y.; Chen, L.; Gu, L.; Zhang, F.; Ouyang, J.; Zhang, X. GSTA1 expression is correlated with aldosterone level in KCNJ5-mutated adrenal aldosterone-producing adenoma. J. Clin. Endocrinol. Metab. 2018, 103, 813–823.
- Der Teo, A.E.; Garg, S.; Johnson, T.I.; Zhao, W.; Zhou, J.; Gomez-Sanchez, C.E.; Gurnell, M.; Brown, M.J. Physiological and Pathological Roles in Human Adrenal of the Glomeruli-Defining Matrix Protein NPNT (Nephronectin). Hypertension 2017, 69, 1207–1216.
- Itcho, K.; Oki, K.; Gomez-Sanchez, C.E.; Gomez-Sanchez, E.P.; Ohno, H.; Kobuke, K.; Nagano, G.; Yoshii, Y.; Baba, R.; Hattori, N.; et al. Endoplasmic Reticulum Chaperone Calmegin Is Upregulated in Aldosterone-Producing Adenoma and Associates with Aldosterone Production. Hypertension 2020, 75, 492–499.
- Howard, B.; Wang, Y.; Xekouki, P.; Faucz, F.R.; Jain, M.; Zhang, L.; Meltzer, P.G.; Stratakis, C.A.; Kebebew, E. Integrated analysis of genome-wide methylation and gene expression shows epigenetic regulation of CYP11B2 in aldosteronomas. J. Clin. Endocrinol. Metab. 2014, 99, 536–543.
- Murakami, M.; Yoshimoto, T.; Nakabayashi, K.; Tsuchiya, K.; Minami, I.; Bouchi, R.; Izumiyama, H.; Fujii, Y.; Abe, K.; Tayama, C.; et al. Integration of transcriptome and methylome analysis of aldosterone-producing adenomas. Eur. J. Endocrinol. 2015, 173, 185–195.
- Yoshii, Y.; Oki, K.; Gomez-Sanchez, C.E.; Ohno, H.; Itcho, K.; Kobuke, K.; Yoneda, M. Hypomethylation of CYP11B2 in Aldosterone-Producing Adenoma. Hypertension 2016, 68, 1432–1437.
- Di Dalmazi, G.; Morandi, L.; Rubin, B.; Pilon, C.; Asioli, S.; Vicennati, V.; De Leo, A.; Ambrosi, F.; Santini, D.; Pagotto, U.; et al. DNA Methylation of Steroidogenic Enzymes in Benign Adrenocortical Tumors: New Insights in Aldosterone-Producing Adenomas. J. Clin. Endocrinol. Metab. 2020, 105, 1–11.
- Sutherland, D.J.; Ruse, J.L.; Laidlaw, J.C. Hypertension, increased aldosterone secretion and low plasma renin activity relieved by dexamethasone. Can. Med. Assoc. J. 1966, 95, 1109–1119.
- Lifton, R.P.; Dluhy, R.G.; Powers, M.; Rich, G.M.; Cook, S.; Ulick, S.; Lalouel, J.-M. A chimaeric llβ-hydroxylase/aldosterone synthase gene causes glucocorticoid-remediable aldosteronism and human hypertension. Nature 1992, 355, 262–265.
- Carroll, J.; Dluhy, R.; Fallo, F.; Pistorello, M.; Bradwin, G.; Gomez-Sanchez, C.E.; Mortensen, R. Aldosterone-producing adenomas do not contain glucocorticoid-remediable aldosteronism chimeric gene duplications. J. Clin. Endocrinol. Metab. 1996, 81, 4310–4312.
- Oki, K.; Plonczynski, M.W.; Lam, M.L.; Gomez-Sanchez, E.P.; Gomez-Sanchez, C.E. Potassium channel mutant KCNJ5 T158A expression in HAC-15 cells increases aldosterone synthesis. Endocrinology 2012, 153, 1774–1782.
- Hattangady, N.G.; Karashima, S.; Yuan, L.; Ponce-Balbuena, D.; Jalife, J.; Gomez-Sanchez, C.E.; Auchus, R.J.; Rainey, W.E.; Else, T. Mutated KCNJ5 activates the acute and chronic regulatory steps in aldosterone production. J. Mol. Endocrinol. 2016, 57, 1–11.
- Tauber, P.; Penton, D.; Stindl, J.; Humberg, E.; Tegtmeier, I.; Sterner, C.; Beuschlein, F.; Reincke, M.; Barhanin, J.; Bandulik, S.; et al. Pharmacology and pathophysiology of mutated KCNJ5 found in adrenal aldosterone-producing adenomas. Endocrinology 2014, 155, 1353–1362.
- Kuppusamy, M.; Caroccia, B.; Stindl, J.; Bandulik, S.; Lenzini, L.; Gioco, F.; Fishman, V.; Zanotti, G.; Gomez-Sanchez, C.; Bader, M.; et al. A novel KCNJ5-insT149 somatic mutation close to, but outside, the selectivity filter causes resistant hypertension by loss of selectivity for potassium. J. Clin. Endocrinol. Metab. 2014, 99, E1765–E1773.
- Monticone, S.; Hattangady, N.G.; Nishimoto, K.; Mantero, F.; Rubin, B.; Cicala, M.V.; Pezzani, R.; Auchus, R.J.; Ghayee, H.K.; Shibata, H.; et al. Effect of KCNJ5 mutations on gene expression in aldosterone-producing adenomas and adrenocortical cells. J. Clin. Endocrinol. Metab. 2012, 97, 1567–1572.
- Oki, K.; Gomez-Sanchez, C.E. The landscape of molecular mechanism for aldosterone production in aldosterone-producing adenoma. Endocr. J. 2020, 67, 989–995.
- Boulkroun, S.; Golib Dzib, J.F.; Samson-Couterie, B.; Rosa, F.L.F.; Rickard, A.J.; Meatchi, T.; Amar, L.; Benecke, A.; Zennaro, M.C. KCNJ5 mutations in aldosterone producing adenoma and relationship with adrenal cortex remodeling. Mol. Cell. Endocrinol. 2013, 371, 221–227.
- Yang, Y.; Gomez-Sanchez, C.E.; Jaquin, D.; Aristizabal Prada, E.T.; Meyer, L.S.; Knösel, T.; Schneider, H.; Beuschlein, F.; Reincke, M.; Williams, T.A. Primary Aldosteronism: KCNJ5 Mutations and Adrenocortical Cell Growth. Hypertension 2019, 74, 809–816.
- Cheng, C.J.; Sung, C.C.; Wu, S.T.; Lin, Y.C.; Sytwu, H.K.; Huang, C.L.; Lin, S.H. Novel KCNJ5 mutations in sporadic aldosterone-producing adenoma reduce Kir3.4 membrane abundance. J. Clin. Endocrinol. Metab. 2015, 100, E155–E163.
- Williams, T.A.; Monticone, S.; Schack, V.R.; Stindl, J.; Burrello, J.; Buffolo, F.; Annaratone, L.; Castellano, I.; Beuschlein, F.; Reincke, M.; et al. Somatic ATP1A1, ATP2B3, and KCNJ5 mutations in aldosterone-producing adenomas. Hypertension 2014, 63, 188–195.
- Hardege, I.; Xu, S.; Gordon, R.D.; Thompson, A.J.; Figg, N.; Stowasser, M.; Murrell-Lagnado, R.; O’Shaughnessy, K.M. Novel insertion mutation in KCNJ5 channel produces constitutive aldosterone release from H295R cells. Mol. Endocrinol. 2015, 29, 1522–1530.
- Nanba, K.; Omata, K.; Else, T.; Beck, P.C.C.; Nanba, A.T.; Turcu, A.F.; Miller, B.S.; Giordano, T.J.; Tomlins, S.A.; Rainey, W.E. Targeted molecular characterization of aldosterone-producing adenomas in white americans. J. Clin. Endocrinol. Metab. 2018, 103, 3869–3876.
- Scholl, U.I.; Healy, J.M.; Thiel, A.; Fonseca, A.L.; Brown, T.C.; Kunstman, J.W.; Horne, M.J.; Dietrich, D.; Riemer, J.; Kücükköylü, S.; et al. Novel somatic mutations in primary hyperaldosteronism are related to the clinical, radiological and pathological phenotype. Clin. Endocrinol. 2015, 83, 779–789.
- Åkerström, T.; Crona, J.; Delgado Verdugo, A.; Starker, L.F.; Cupisti, K.; Willenberg, H.S.; Knoefel, W.T.; Saeger, W.; Feller, A.; Ip, J.; et al. Comprehensive re-sequencing of adrenal aldosterone producing lesions reveal three somatic mutations near the KCNJ5 potassium channel selectivity filter. PLoS ONE 2012, 7, e41926.
- Nanba, K.; Omata, K.; Tomlins, S.A.; Giordano, T.J.; Hammer, G.D.; Rainey, W.E.; Else, T. Double adrenocortical adenomas harboring independent KCNJ5 and PRKACA somatic mutations. Eur. J. Endocrinol. 2016, 175, K1–K6.
- Fang-Fang, Z.; Li-Min, Z.; Ai-Fang, N.; Xiao-Ying, L.; Jing-Rong, L.; Ke, Z.; Jing, C.; Wen-Long, Z.; Zhou-Jun, S.; Yi-Chun, Z.; et al. Clinical Characteristics of Somatic Mutations in Chinese Patients With Aldosterone-Producing Adenoma. Hypertension 2015, 65, 622–628.
- Nanba, K.; Omata, K.; Gomez-Sanchez, C.E.; Stratakis, C.A.; Demidowich, A.P.; Suzuki, M.; Thompson, L.D.R.; Cohen, D.L.; Luther, J.M.; Gellert, L.; et al. Genetic characteristics of aldosterone-producing adenomas in blacks. Hypertension 2019, 73, 885–892.
- Kitamoto, T.; Omura, M.; Suematsu, S.; Saito, J.; Nishikawa, T. KCNJ5 mutation as a predictor for resolution of hypertension after surgical treatment of aldosterone-producing adenoma. J. Hypertens. 2018, 36, 619–627.
- Azizan, E.A.B.; Murthy, M.; Stowasser, M.; Gordon, R.; Kowalski, B.; Xu, S.; Brown, M.J.; O’Shaughnessy, K.M. Somatic mutations affecting the selectivity filter of KCNJ5 are frequent in 2 large unselected collections of adrenal aldosteronomas. Hypertension 2012, 59, 587–591.
- Fernandes-Rosa, F.L.; Williams, T.A.; Riester, A.; Steichen, O.; Beuschlein, F.; Boulkroun, S.; Strom, T.M.; Monticone, S.; Amar, L.; Meatchi, T.; et al. Genetic spectrum and clinical correlates of somatic mutations in aldosterone-producing adenoma. Hypertension 2014, 64, 354–361.
- Taguchi, R.; Yamada, M.; Nakajima, Y.; Satoh, T.; Hashimoto, K.; Shibusawa, N.; Ozawa, A.; Okada, S.; Rokutanda, N.; Takata, D.; et al. Expression and mutations of KCNJ5 mRNA in Japanese patients with aldosterone-producing adenomas. J. Clin. Endocrinol. Metab. 2012, 97, 1311–1319.
- Ram Hong, A.; Kim, J.H.; Song, Y.S.; Lee, K.E.; Seo, S.H.; Seong, M.W.; Shin, C.S.; Kim, S.W.; Kim, S.Y. Genetics of aldosterone-producing adenoma in Korean patients. PLoS ONE 2016, 11, e0147590.
- Okamura, T.; Nakajima, Y.; Katano-Toki, A.; Horiguchi, K.; Matsumoto, S.; Yoshino, S.; Yamada, E.; Tomaru, T.; Ishii, S.; Saito, T.; et al. Characteristics of japanese aldosterone-producing adenomas with KCNJ5 mutations. Endocr. J. 2017, 64, 39–47.
- Warachit, W.; Atikankul, T.; Houngngam, N.; Sunthornyothin, S. Prevalence of somatic KCNJ5 mutations in Thai patients with aldosterone-producing adrenal adenomas. J. Endocr. Soc. 2018, 2, 1137–1146.
- Wu, V.C.; Huang, K.H.; Peng, K.Y.; Tsai, Y.C.; Wu, C.H.; Wang, S.M.; Yang, S.Y.; Lin, L.Y.; Chang, C.C.; Lin, Y.H.; et al. Prevalence and clinical correlates of somatic mutation in aldosterone producing adenoma-Taiwanese population. Sci. Rep. 2015, 5, 1–10.
- Lenzini, L.; Rossitto, G.; Maiolino, G.; Letizia, C.; Funder, J.W.; Rossi, G.P. A meta-analysis of somatic KCNJ5 K+ channel mutations in 1636 patients with an aldosterone-producing adenoma. J. Clin. Endocrinol. Metab. 2015, 100, E1089–E1095.
- Yamada, M.; Nakajima, Y.; Taguchi, R.; Okamura, T.; Ishii, S.; Tomaru, T.; Ozawa, A.; Shibusawa, N.; Yoshino, S.; Toki, A.; et al. KCNJ5 mutations in aldosterone- and cortisol-co-secreting adrenal adenomas. Endocr. J. 2012, 59, 735–741.
- Peng, K.Y.; Liao, H.W.; Chan, C.K.; Lin, W.C.; Yang, S.Y.; Tsai, Y.C.; Huang, K.H.; Lin, Y.H.; Chueh, J.S.; Wu, V.C. Presence of subclinical hypercortisolism in clinical aldosterone-producing adenomas predicts lower clinical success. Hypertension 2020, 76, 1537–1544.
- Rossi, G.P.; Cesari, M.; Letizia, C.; Seccia, T.M.; Cicala, M.V.; Zinnamosca, L.; Kuppusamy, M.; Mareso, S.; Sciomer, S.; Iacobone, M.; et al. KCNJ5 gene somatic mutations affect cardiac remodelling but do not preclude cure of high blood pressure and regression of left ventricular hypertrophy in primary aldosteronism. J. Hypertens. 2014, 32, 1514–1522.
- Kitamoto, T.; Suematsu, S.; Matsuzawa, Y.; Saito, J.; Omura, M.; Nishikawa, T. Comparison of cardiovascular complications in patients with and without KCNJ5 gene mutations harboring aldosterone-producing adenomas. J. Atheroscler. Thromb. 2015, 22, 191–200.
- Chang, Y.-Y.; Tsai, C.-H.; Peng, S.-Y.; Chen, Z.-W.; Chang, C.-C.; Lee, B.-C.; Liao, C.-W.; Pan, C.-T.; Chen, Y.-L.; Lin, L.-C.; et al. KCNJ5 Somatic Mutations in Aldosterone-Producing Adenoma Are Associated With a Worse Baseline Status and Better Recovery of Left Ventricular Remodeling and Diastolic Function. Hypertension 2021, 77, 114–125.
- Vilela, L.A.P.; Rassi-Cruz, M.; Guimaraes, A.G.; Moises, C.C.S.; Freitas, T.C.; Alencar, N.P.; Petenuci, J.; Goldbaum, T.S.; MacIel, A.A.W.; Pereira, M.A.A.; et al. KCNJ5 Somatic Mutation Is a Predictor of Hypertension Remission after Adrenalectomy for Unilateral Primary Aldosteronism. J. Clin. Endocrinol. Metab. 2019, 104, 4695–4702.
- Nanba, K.; Chen, A.X.; Omata, K.; Vinco, M.; Giordano, T.J.; Else, T.; Hammer, G.D.; Tomlins, S.A.; Rainey, W.E. Molecular heterogeneity in aldosterone-producing adenomas. J. Clin. Endocrinol. Metab. 2016, 101, 999–1007.
- Azizan, E.A.B.; Lam, B.Y.H.; Newhouse, S.J.; Zhou, J.; Kuc, R.E.; Clarke, J.; Happerfield, L.; Marker, A.; Hoffman, G.J.; Brown, M.J. Microarray, qPCR, and KCNJ5 sequencing of aldosterone-producing adenomas reveal differences in genotype and phenotype between zona glomerulosa- and zona fasciculata-like tumors. J. Clin. Endocrinol. Metab. 2012, 97, 819–829.
- Inoue, K.; Yamazaki, Y.; Kitamoto, T.; Hirose, R.; Saito, J.; Omura, M.; Sasano, H.; Nishikawa, T. Aldosterone Suppression by Dexamethasone in Patients with KCNJ5-Mutated Aldosterone-Producing Adenoma. J. Clin. Endocrinol. Metab. 2018, 103, 3477–3485.
- Ono, Y.; Yamazaki, Y.; Omata, K.; Else, T.; Tomlins, S.A.; Rhayem, Y.; Williams, T.A.; Reincke, M.; Carling, T.; Monticone, S.; et al. Histological Characterization of Aldosterone-producing Adrenocortical Adenomas with Different Somatic Mutations. J. Clin. Endocrinol. Metab. 2020, 105, e282–e289.
- Williams, T.A.; Peitzsch, M.; Dietz, A.S.; Dekkers, T.; Bidlingmaier, M.; Riester, A.; Treitl, M.; Rhayem, Y.; Beuschlein, F.; Lenders, J.W.M.; et al. Genotype-Specific Steroid Profiles Associated With Aldosterone-Producing Adenomas. Hypertension 2016, 67, 139–145.
- Tezuka, Y.; Yamazaki, Y.; Kitada, M.; Morimoto, R.; Kudo, M.; Seiji, K.; Takase, K.; Kawasaki, Y.; Mitsuzuka, K.; Ito, A.; et al. 18-Oxocortisol Synthesis in Aldosterone-Producing Adrenocortical Adenoma and Significance of KCNJ5 Mutation Status. Hypertension 2019, 73, 1283–1290.
- Satoh, F.; Morimoto, R.; Ono, Y.; Iwakura, Y.; Omata, K.; Kudo, M.; Takase, K.; Seiji, K.; Sasamoto, H.; Honma, S.; et al. Measurement of peripheral plasma 18-oxocortisol can discriminate unilateral adenoma from bilateral diseases in patients with primary aldosteronism. Hypertension 2015, 65, 1096–1102.
- Eisenhofer, G.; Dekkers, T.; Peitzsch, M.; Dietz, A.S.; Bidlingmaier, M.; Treitl, M.; Williams, T.A.; Bornstein, S.R.; Haase, M.; Rump, L.C.; et al. Mass spectrometry-based adrenal and peripheral venous steroid profiling for subtyping primary aldosteronism. Clin. Chem. 2016, 62, 514–524.
- Turcu, A.F.; Wannachalee, T.; Tsodikov, A.; Nanba, A.T.; Ren, J.; Shields, J.J.; O’Day, P.J.; Giacherio, D.; Rainey, W.E.; Auchus, R.J. Comprehensive Analysis of Steroid Biomarkers for Guiding Primary Aldosteronism Subtyping. Hypertension 2020, 75, 183–192.
- Eisenhofer, G.; Durán, C.; Cannistraci, C.V.; Peitzsch, M.; Williams, T.A.; Riester, A.; Burrello, J.; Buffolo, F.; Prejbisz, A.; Beuschlein, F.; et al. Use of Steroid Profiling Combined With Machine Learning for Identification and Subtype Classification in Primary Aldosteronism. JAMA Netw. Open 2020, 3, e2016209.
- Geller, D.S.; Zhang, J.; Wisgerhof, M.V.; Shackleton, C.; Kashgarian, M.; Lifton, R.P. A novel form of human mendelian hypertension featuring nonglucocorticoid- remediable aldosteronism. J. Clin. Endocrinol. Metab. 2008, 93, 3117–3123.
- Charmandari, E.; Sertedaki, A.; Kino, T.; Merakou, C.; Hoffman, D.A.; Hatch, M.M.; Hurt, D.E.; Lin, L.; Xekouki, P.; Stratakis, C.A.; et al. A novel point mutation in the KCNJ5 gene causing primary hyperaldosteronism and early-onset autosomal dominant hypertension. J. Clin. Endocrinol. Metab. 2012, 97, 1532–1539.
- Monticone, S.; Bandulik, S.; Stindl, J.; Zilbermint, M.; Dedov, I.; Mulatero, P.; Allgaeuer, M.; Lee, C.C.R.; Stratakis, C.A.; Williams, T.A.; et al. A case of severe hyperaldosteronism caused by a de novo mutation affecting a critical salt bridge Kir3.4 residue. J. Clin. Endocrinol. Metab. 2015, 100, E114–E118.
- Scholl, U.I.; Nelson-Williams, C.; Yue, P.; Grekin, R.; Wyatt, R.J.; Dillon, M.J.; Couch, R.; Hammer, L.K.; Harley, F.L.; Farhi, A.; et al. Hypertension with or without adrenal hyperplasia due to different inherited mutations in the potassium channel KCNJ5. Proc. Natl. Acad. Sci. USA 2012, 109, 2533–2538.
- Mulatero, P.; Tauber, P.; Zennaro, M.C.; Monticone, S.; Lang, K.; Beuschlein, F.; Fischer, E.; Tizzani, D.; Pallauf, A.; Viola, A.; et al. KCNJ5 mutations in European families with nonglucocorticoid remediable familial hyperaldosteronism. Hypertension 2012, 59, 235–240.
- Monticone, S.; Hattangady, N.G.; Penton, D.; Isales, C.M.; Edwards, M.A.; Williams, T.A.; Sterner, C.; Warth, R.; Mulatero, P.; Rainey, W.E. A novel Y152C KCNJ5 mutation responsible for familial hyperaldosteronism type III. J. Clin. Endocrinol. Metab. 2013, 98, 1861–1865.
- Adachi, M.; Muroya, K.; Asakura, Y.; Sugiyama, K.; Homma, K.; Hasegawa, T. Discordant genotype-phenotype correlation in familial hyperaldosteronism type III with KCNJ5 gene mutation: A patient report and review of the literature. Horm. Res. Paediatr. 2014, 82, 138–142.
- Tamura, A.; Nishimoto, K.; Seki, T.; Matsuzawa, Y.; Saito, J.; Omura, M.; Gomez-Sanchez, C.E.; Makita, K.; Matsui, S.; Moriya, N.; et al. Somatic KCNJ5 mutation occurring early in adrenal development may cause a novel form of juvenile primary aldosteronism. Mol. Cell. Endocrinol. 2017, 441, 134–139.
- Maria, A.G.; Suzuki, M.; Berthon, A.; Kamilaris, C.; Demidowich, A.; Lack, J.; Zilbermint, M.; Hannah-Shmouni, F.; Faucz, F.R.; Stratakis, C.A. Mosaicism for KCNJ5 Causing Early-Onset Primary Aldosteronism due to Bilateral Adrenocortical Hyperplasia. Am. J. Hypertens. 2020, 33, 124–130.
- Stindl, J.; Tauber, P.; Sterner, C.; Tegtmeier, I.; Warth, R.; Bandulik, S. Pathogenesis of adrenal aldosterone-producing adenomas carrying mutations of the Na+/K+-ATPase. Endocrinology 2015, 156, 4582–4591.
- Nanba, K.; Yamazaki, Y.; Bick, N.; Onodera, K.; Tezuka, Y.; Omata, K.; Ono, Y.; Blinder, A.R.; Tomlins, S.A.; Rainey, W.E.; et al. Prevalence of Somatic Mutations in Aldosterone-Producing Adenomas in Japanese Patients. J. Clin. Endocrinol. Metab. 2020, 105, 1–8.
- De Sousa, K.; Boulkroun, S.; Baron, S.; Nanba, K.; Wack, M.; Rainey, W.E.; Rocha, A.; Giscos-Douriez, I.; Meatchi, T.; Amar, L.; et al. Genetic, cellular, and molecular heterogeneity in adrenals with aldosterone-producing adenoma. Hypertension 2020, 75, 1034–1044.
- Kitamoto, T.; Suematsu, S.; Yamazaki, Y.; Nakamura, Y.; Sasano, H.; Matsuzawa, Y.; Saito, J.; Omura, M.; Nishikawa, T. Clinical and steroidogenic characteristics of aldosterone-producing adenomas with ATPase or CACNA1D gene mutations. J. Clin. Endocrinol. Metab. 2016, 101, 494–503.
- Backman, S.; Åkerström, T.; Maharjan, R.; Cupisti, K.; Willenberg, H.S.; Hellman, P.; Björklund, P. RNA Sequencing Provides Novel Insights into the Transcriptome of Aldosterone Producing Adenomas. Sci. Rep. 2019, 9, 1–10.
- Tauber, P.; Aichinger, B.; Christ, C.; Stindl, J.; Rhayem, Y.; Beuschlein, F.; Warth, R.; Bandulik, S. Cellular pathophysiology of an adrenal adenoma-associated mutant of the plasma membrane Ca2+-ATPase ATP2B3. Endocrinology 2016, 157, 2489–2499.
- Dekkers, T.; Ter Meer, M.; Lenders, J.W.M.; Hermus, A.R.M.; Schultze Kool, L.; Langenhuijsen, J.F.; Nishimoto, K.; Ogishima, T.; Mukai, K.; Azizan, E.A.B.; et al. Adrenal nodularity and somatic mutations in primary aldosteronism: One node is the culprit? J. Clin. Endocrinol. Metab. 2014, 99, 1341–1351.
- Ortner, N.J.; Kaserer, T.; Copeland, J.N.; Striessnig, J. De novo CACAN1D Ca2+ channelopathies: Clinical phenotypes and molecular mechanism. Pflug. Arch. Eur. J. Physiol. 2020, 472, 755–773.
- Semenova, N.A.; Ryzhkova, O.R.; Strokova, T.V.; Taran, N.N. The third case report a patient with primary aldosteronism, seizures, and neurologic abnormalities (PASNA) syndrome de novo variant mutations in the CACNA1D gene. Zhurnal Nevrol. Psikhiatrii Im. S.S. Korsakova 2018, 118, 49–52.
- De Mingo Alemany, M.C.; Mifsud Grau, L.; Moreno Macián, F.; Ferrer Lorente, B.; León Cariñena, S. A de novo CACNA1D missense mutation in a patient with congenital hyperinsulinism, primary hyperaldosteronism and hypotonia. Channels 2020, 14, 175–180.
- Kim, A.C.; Reuter, A.L.; Zubair, M.; Else, T.; Serecky, K.; Bingham, N.C.; Lavery, G.G.; Parker, K.L.; Hammer, G.D. Targeted disruption β-catenin in Sf1-expressing cells impairs development and maintenance of the adrenal cortex. Development 2008, 135, 2593–2602.
- Boulkroun, S.; Samson-Couterie, B.; Golib-Dzib, J.F.; Amar, L.; Plouin, P.F.; Sibony, M.; Lefebvre, H.; Louiset, E.; Jeunemaitre, X.; Meatchi, T.; et al. Aldosterone-producing adenoma formation in the adrenal cortex involves expression of stem/progenitor cell markers. Endocrinology 2011, 152, 4753–4763.
- Berthon, A.; Drelon, C.; Ragazzon, B.; Boulkroun, S.; Tissier, F.; Amar, L.; Samson-Couterie, B.; Zennaro, M.C.; Plouin, P.F.; Skah, S.; et al. WNT/β-catenin signalling is activated in aldosteroneproducing adenomas and controls aldosterone production. Hum. Mol. Genet. 2014, 23, 889–905.
- Wu, V.C.; Wang, S.M.; Chueh, S.C.J.; Yang, S.Y.; Huang, K.H.; Lin, Y.H.; Wang, J.J.; Connolly, R.; Hu, Y.H.; Gomez-Sanchez, C.E.; et al. The prevalence of CTNNB1 mutations in primary aldosteronism and consequences for clinical outcomes. Sci. Rep. 2017, 7, 1–10.
- Tissier, F.; Cavard, C.; Groussin, L.; Perlemoine, K.; Fumey, G.; Hagneré, A.M.; René-Corail, F.; Jullian, E.; Gicquel, C.; Bertagna, X.; et al. Mutations of β-catenin in adrenocortical tumors: Activation of the Wnt signaling pathway is a frequent event in both benign and malignant adrenocortical tumors. Cancer Res. 2005, 65, 7622–7627.
- Berthon, A.; Sahut-Barnola, I.; Lambert-Langlais, S.; de Joussineau, C.; Damon-Soubeyrand, C.; Louiset, E.; Taketo, M.M.; Tissier, F.; Bertherat, J.; Lefrançois-Martinez, A.M.; et al. Constitutive β-catenin activation induces adrenal hyperplasia and promotes adrenal cancer development. Hum. Mol. Genet. 2010, 19, 1561–1576.
- Gordon, R.D.; Stowasser, M.; Tunny, T.J.; Klemm, S.A.; Finn, W.L.; Krek, A.L. Clinical and pathological diversity of primary aldosteronism, including a new familial variety. Clin. Exp. Pharmacol. Physiol. 1991, 18, 283–286.
- Stowasser, M.; Gordon, R.D.; Tunny, T.J.; Klemm, S.A.; Finn, W.L.; Krek, A.L. Familial hyperaldosteronism type II: Five families with a new variety of primary aldosteronism. Clin. Exp. Pharmacol. Physiol. 1992, 19, 319–322.
- Dutta, R.K.; Arnesen, T.; Heie, A.; Walz, M.; Alesina, P.; Söderkvist, P.; Gimm, O. A somatic mutation in CLCN2 identified in a sporadic aldosterone-producing adenoma. Eur. J. Endocrinol. 2019, 181, K37–K41.
- Rege, J.; Nanba, K.; Blinder, A.R.; Plaska, S.; Udager, A.M.; Vats, P.; Kumar-Sinha, C.; Giordano, T.J.; Rainey, W.E.; Else, T. Identification of Somatic Mutations in CLCN2 in Aldosterone-Producing Adenomas. J. Endocr. Soc. 2020, 4, bvaa123.
- Daniil, G.; Fernandes-Rosa, F.L.; Chemin, J.; Blesneac, I.; Beltrand, J.; Polak, M.; Jeunemaitre, X.; Boulkroun, S.; Amar, L.; Strom, T.M.; et al. CACNA1H Mutations Are Associated With Different Forms of Primary Aldosteronism. EBioMedicine 2016, 13, 225–236.
- Nanba, K.; Blinder, A.R.; Rege, J.; Hattangady, N.G.; Else, T.; Liu, C.J.; Tomlins, S.A.; Vats, P.; Kumar-Sinha, C.; Giordano, T.J.; et al. Somatic CACNA1H Mutation As a Cause of Aldosterone-Producing Adenoma. Hypertension 2020, 75, 645–649.
- Beuschlein, F.; Fassnacht, M.; Assié, G.; Calebiro, D.; Stratakis, C.A.; Osswald, A.; Ronchi, C.L.; Wieland, T.; Sbiera, S.; Faucz, F.R.; et al. Constitutive Activation of PKA Catalytic Subunit in Adrenal Cushing’s Syndrome. N. Engl. J. Med. 2014, 370, 1019–1028.
- Rhayem, Y.; Perez-Rivas, L.G.; Dietz, A.; Bathon, K.; Gebhard, C.; Riester, A.; Mauracher, B.; Gomez-Sanchez, C.; Eisenhofer, G.; Schwarzmayr, T.; et al. PRKACA somatic mutations are rare findings in aldosterone-producing adenomas. J. Clin. Endocrinol. Metab. 2016, 101, 3010–3017.
- Nakajima, Y.; Okamura, T.; Horiguchi, K.; Gohko, T.; Miyamoto, T.; Satoh, T.; Ozawa, A.; Ishii, S.; Yamada, E.; Hashimoto, K.; et al. Gnas mutations in adrenal aldosterone-producing adenomas. Endocr. J. 2016, 63, 199–204.
- Zilbermint, M.; Xekouki, P.; Faucz, F.R.; Berthon, A.; Gkourogianni, A.; Schernthaner-Reiter, M.H.; Batsis, M.; Sinaii, N.; Quezado, M.M.; Merino, M.; et al. Primary aldosteronism and ARMC5 variants. J. Clin. Endocrinol. Metab. 2015, 100, E900–E909.
- Mulatero, P.; Schiavi, F.; Williams, T.A.; Monticone, S.; Barbon, G.; Opocher, G.; Fallo, F. ARMC5 mutation analysis in patients with primary aldosteronism and bilateral adrenal lesions. J. Hum. Hypertens. 2016, 30, 374–378.
- Joseph, J.J.; Zhou, X.; Zilbermint, M.; Stratakis, C.A.; Faucz, F.R.; Lodish, M.B.; Berthon, A.; Wilson, J.G.; Hsueh, W.A.; Golden, S.H.; et al. The Association of ARMC5 with the Renin-Angiotensin-Aldosterone System, Blood Pressure, and Glycemia in African Americans. J. Clin. Endocrinol. Metab. 2020, 105, 2625–2633.
- Hattangady, N.G.; Foster, J.; Lerario, A.M.; Ponce-Balbuena, D.; Rege, J.; Monticone, S.; Rainey, W.E.; Mulatero, P.; Else, T. Molecular and Electrophysiological Analyses of ATP2B4 Gene Variants in Bilateral Adrenal Hyperaldosteronism. Horm. Cancer 2020, 11, 52–62.
- Rassi-Cruz, M.; Maria, A.G.; Faucz, F.R.; London, E.; Vilela, L.A.P.; Santana, L.S.; Benedetti, A.F.F.; Goldbaum, T.S.; Tanno, F.Y.; Srougi, V.; et al. Phosphodiesterase 2A and 3B variants are associated with primary aldosteronism. Endocr. Relat. Cancer 2021, 28, 1–13.