 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Cristina Nativi | + 6696 word(s) | 6696 | 2021-04-22 09:20:39 | | | |
| 2 | Conner Chen | Meta information modification | 6696 | 2021-04-29 08:59:02 | | |
Video Upload Options
Vaccines are the most effective medical intervention due to their continual success in preventing infections and improving mortality worldwide. Early vaccines were developed empirically however, rational design of vaccines can allow us to optimise their efficacy, by tailoring the immune response. Establishing the immune correlates of protection greatly informs the rational design of vaccines. This facilitates the selection of the best vaccine antigens and the most appropriate vaccine adjuvant to generate optimal memory immune T cell and B cell responses. This review outlines the range of vaccine types that are currently authorised and those under development. We outline the optimal immunological correlates of protection that can be targeted.
1. The Immunological Correlate(s) of Protection
Before embarking on the design of a vaccine it is very important to first consider what type of immune response will generate the desired vaccine-induced immunity. While this may seem obvious, this approach was not followed for many existing vaccines developed for infectious diseases in the last century. As mentioned, many current vaccines have been developed empirically and the immunological correlates of protection are not yet clearly defined for many infectious diseases.
Historically most vaccines aimed to induce neutralizing antibody responses. Neutralizing antibodies are relatively easy to measure in the serum of immunized individuals and the detection of neutralizing antibodies in serum samples likely reflects their activity in the infected host. However, numerous infectious diseases and malignancies cannot be prevented or cured (in case of a therapeutic vaccine) by neutralizing antibodies. In most cases these infections show an intracellular life cycle, or a high degree of a variability of surface antigens, which cannot be targeted since they escape from vaccine-induced antibodies. Such diseases, if preventable at all, often require cell-mediated or cellular immunity, preferably directed against a conserved non-variable antigen(s), to impede the disease. As a result, many vaccine approaches have failed due to their inadequate design and inability to induce a cell-mediated immune response.
1.1. Passive Immunity Transfer or Immunity Depletion
Knowledge of the contribution of specific immune effector mechanisms involved in protection against a particular pathogen or tumor, will help to identify the type of immune response that should be evoked by effectively-designed vaccines. In order to identify which type of immune reaction is necessary for protection against a particular disease a range of approaches can be followed. Animal models can be used to decipher the contribution of antibodies or specific immune cell populations. One strategy involves the assessment of passive protection of either antibodies or immune T cells from immune donors to naïve recipients that are subsequently exposed to an artificial challenge with the pathogen or tumor of interest. Another approach involves the selective depletion of either antibody-producing B cells or T cells (or their CD4 or CD8 subpopulations) in immune hosts and the subsequent monitoring of a drop in protective immune function after challenge. The contribution of antibody-producing B cells can be studied by using B cell-neutralizing antibodies or by specific genetic deletion of the B cell population (e.g., μMT gene deficient mice). Alternatively, the contribution of T cells to immunity in immune hosts can be examined using T cell-depleting antibodies, or mice with genetically deleted T cells (or their subpopulations). Any reduction in immunity relative to wild-type or depleted immune mouse models indicate their contribution.
Despite these experimental strategies, the specific contributions of distinct immune effector mechanisms are known for just a few pathogens. This may be due in part to the lack of suitable animal models reflecting human or veterinary disease, the complex changes in life cycle of certain pathogens and their evasion from immune surveillance, as well as the complex nature of immune reactions and synergistic activities of immune cell populations. For example, antibodies may be involved in protection during the acute phase of infection while cell-mediated responses may become critical during the chronic phase of infection. Although animal models may not fully reflect the disease or may not be available, in many cases there are clues to the signature of immune clearance mechanisms that are associated with recovery from, or immunity to, specific infectious diseases. Extrapolations to correlates of immunity can be made based on knowledge from related categories of intracellular versus extracellular pathogens, or related types of micro-organisms with similar life cycles. Actual correlates have been defined or suggested for Hib, pneumococcal and meningococcal vaccines. Moreover, for most tumors we know that in general type-1 immune and STING pathway interferon responses are related to protection [1]. Hence, the immune correlate of protection can be estimated or is predictable to some extend without the need for immunity transfer or depletion experiments.
The systems biology approach can facilitate a better picture of the immune responses in general and to vaccination in humans [2]; in fact, it may prove that vaccination-induced artificial immune effector clearance, rather than natural immune effector responses, may prove sufficient to fight of particular pathogens or tumors and may provide even better levels of protection. Understanding the immunological mechanisms of vaccination and the bridging of technical knowledge gaps [3] will help in the rational design of future vaccines against emerging infectious pathogens, such as COVID-19, as well as against prominent global diseases such as HIV, malaria, and tuberculosis.
1.2. Types of Protective Immune Responses
1.2.1. Antibody-Based Immunity
Humoral or antibody-based immunity is one of the two arms of the adaptive immune response, which results in the generation of antigen-specific antibodies that target invading microbial pathogens or vaccine antigens. Humoral immunity is achieved by B-cells but requires help from CD4+ T cells and therefore is also dependent on successful cell-mediated immunity. Activated B cells interact with antigen-specific CD4+ helper T cells in the outer cortex of the lymph nodes and undergo proliferation in the presence of cytokines such as IL-4 and IL-5 produced by CD4+ T cells. The antibodies produced bind the organisms and/or their toxins, directly interfering with microbial proliferation via neutralization, opsonization, and complement activation, or will direct other immune cells to phagocytose and destroy the bound microbe. Following a successful induction of humoral immune response, B cells producing affinity matured and isotype-switched antibodies differentiate into quiescent memory B cells (Figure 1).
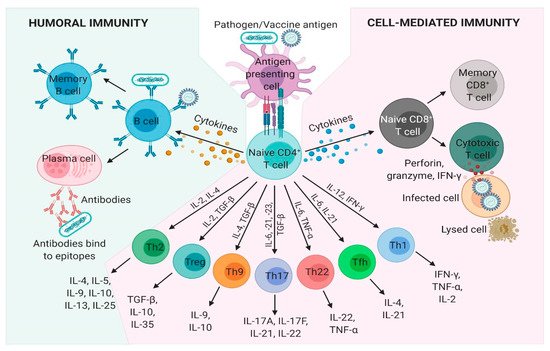
Figure 1. Humoral and cell-mediated adaptive immune responses. Adaptive immunity involves the activation of lymphocytes and develops following exposure to diseases or immunization against diseases through vaccination. Pathogen, tumor or vaccine antigens are recognized by antigen-presenting cells (APCs). Once processed, the antigenic peptides are presented on the surface of major histocompatibility complex (MHC)-II, which are recognized by T cell receptors (TCR) on naïve CD4+ T cells. Upon TCR activation by APCs, naïve CD4+ T cells differentiate into subpopulations: T helper (Th)1, Th2, Th9, Th17, Th22, T follicular helper (Tfh), and T regulatory cells (Treg) under unique cytokine-polarized environments. B-cells, following co-stimulation from cytokines produced by CD4+ T cells, transform into plasma cells that secrete antibodies which circulate in blood and extracellular fluid. A humoral immune response is mediated by secreted antibodies produced by B-cells, which are specific for an individual antigen. CD8+ T cells recognize and bind to intracellularly processed antigenic peptides through their TCR, which are presented on MHC-I molecules on the surface of APCs and infected cells. Cytokines released by CD4+ T cells also stimulate cytotoxic CD8+ T cells, which release effector molecules such as granzyme, perforin, and IFN-γ that destroy infected host cell. A subset of memory B and T cells confer future immunity to the cognate pathogen or antigen. IFN-γ, interferon gamma; IL, interleukin; TGF-β, transforming growth factor β; TNF-α, tumor necrosis factor α.
1.2.2. Cell-Mediated Immunity
Cell-mediated immunity is the other arm of the adaptive immune response that generates a wide variety of antigen specific effector T cells subsets which can either directly kill infected cells or induce various effector functions in conjunction with other immune cells. Naïve CD4+ T cells encounter pathogen or vaccine antigenic peptides complexed with MHC-II molecules that are presented on the surface of antigen presenting cells (APCs), such as dendritic cells and macrophages. Activated APCs provide the critical co-stimulatory signals, which stimulate the T cell receptor (TCR) resulting in T cell proliferation. Following TCR activation by APCs, naïve CD4+ T cells differentiate into either T helper (Th)1, Th2, Th9, Th17, Th22, T follicular helper, and T regulatory cells under unique cytokine-polarized milieus (Figure 1). These subsets of Th cells secrete diverse effector molecules contributing to cell-mediated or humoral immune responses, inflammation or immunoregulation. CD8+ T cells release a variety of cytotoxic molecules such as perforins, granzymes, and IFN-γ, which kill the target cell, e.g., host cells infected with viruses or intracellular bacteria. Once the infection is resolved, antigen-specific CD4+ and CD8+ effector T cells decline in number and a small population is usually maintained as antigen-specific memory CD4+ and CD8+ T cells.
1.2.3. Innate Immunity
Traditionally the evaluation of vaccine efficacy has been based on measuring of specific antibody and T cell readouts or challenge experiments in animal models. However, innate immune activation, which has an important impact on the final outcome of vaccine efficacy and safety, is rarely measured. Early innate immune responses have a well-established role on the eventual down-stream adaptive immunity as well as on early inflammation with potential unwanted side effects of vaccines. It is therefore expected that innate responses may represent a correlate of vaccine efficacy. For many vaccines, the traditional outcome of a vaccination is assessed several weeks after vaccination, however determination of early correlates of vaccine efficacy could speed-up the rational design of vaccines. A well-known problem in evaluating vaccine responses is the large variation in immune responses of the host which is seen in both human and veterinary species. This is driven by heterogeneity in age, genetics, and environmental factors including previous infections, stress or changes in microbiome following antibiotic treatment [4][5][6]. This variation is also a problem in challenge experiments performed for veterinary vaccines and results in poor statistical power of many vaccine trials.
Systems vaccinology provides a solution to these issues. This approach employs multiplexed immune profiling technologies combined with computational modelling to evaluate vaccine responses. This can be applied to peripheral blood samples very early after vaccination, providing molecular signatures of protective immune responses [7]. In fact, it was demonstrated that transcriptomic data can be most informative if analyzed using blood transcriptional modules (BTM) that were created on the basis of highly interacting genes [8][9]. In recent years this approach has continuously been applied to many human studies and, as new technologies have emerged, refined by integration of data from a number of “omics” technologies [10]. In general terms, the data generated using BTM provide information on changes in immune cell population distribution, cellular processes, such as cell cycle and transcription, leukocyte-specific signaling pathways, leukocyte migration, activation of particular immune cell types such as dendritic cells and T cells, inflammation, coagulation, platelet activation, antiviral responses, antigen presentation, immunoglobulin production, or on metabolic processes relevant for immune responses [6][9][11].
Recently such methods have also successfully been adapted to veterinary species including sheep and pigs [12][13] and demonstrated their power to detect and explain immunological processes occurring in tissues such as the injection site of a vaccine using peripheral blood as source. These data can predict vaccine responses and enable a detailed characterization of immune responses induced by different formulations [13]. Interestingly, the correlation patterns of BTM with adaptive responses were seen across multiple species.
In summary, systems vaccinology pipelines can be used to dissect the impact of vaccine components and their formulations on the immune system and thereby help to identify improved delivery systems and immunostimulants. It is possible to identify innate correlates and biomarkers of suitable vaccines to improve formulations, select optimal immunostimulants and compare different batches of vaccines. Finally, such analyses have been used to identify pathways responsible for the heterogeneity in vaccine responses caused by age, nutrition, stress, genetics and the microbiome (reviewed in [10][14]).
2. Choice of Vaccine Antigen
2.1. Rational Antigens for Antibody Immunity
Should immunological data indicate that a disease can be prevented by an antibody response, it is important to select an appropriate antibody-inducing antigen. Induction of robust antibody responses by vaccines requires inclusion of relevant antigenic epitopes for B cells as well as T cell peptide epitopes to elicit appropriate T helper (Th) cell activation and assistance to B cells. Hence, selection of the optimal antigen is imperative in vaccine design.
A successful strategy in the selection of the antigenic determinants as “subunit” vaccines is the isolation and inactivation of essential components of the pathogenic organism [15]. Chemically inactivated toxins isolated from bacteria have been exploited for diphtheria and tetanus vaccines. This approach has also been pursued for subunit vaccines based on purified polysaccharides of Gram-negative bacteria, such as those developed for Streptococcus pneumoniae, Haemophilus influenzae or Salmonella typhi Vi. Saccharide-based vaccines have also been developed in multivalent forms to provide protection against numerous serotypes and serogroups [16].
Current strategies utilize reverse vaccinology approaches to select promising glyco- or protein-derived antigens based on structural studies of the target epitopes of potent antibodies [17][18]. Rational, structure-guided vaccine design can also involve the development of structurally simpler immunogens that present well-defined minimal epitopes targeted by neutralizing antibodies, serving as epitope mimics for elicitation of more focused immune responses [19]. When refined recombinant or synthetic subunit vaccines are designed, the structural features of the antigen may strongly impact the desired antibody-response.
Structural antigen vaccinology is a structural biology approach to design immunogenic antigens. It rationally aims to generate an effective antibody-inducing vaccine antigen, combining experimental methods such as X-ray crystallography, nuclear magnetic resonance (NMR), molecular biology, electron microscopy and mass spectrometry, with computational methods including molecular modeling, virtual screening and epitope prediction [20]. The identification of an antigen candidate is typically based on its cellular location. Although computational approaches exist to predict the protein localization in the cell, a leading technique for antigen identification is mass spectrometry, which allows the assessment of surface structures. Knowing the structure of an antigen will provide important insights into the tertiary structure and position of the potential epitopes. Moreover, structures of antigen-antibody complexes enable the elucidation of the molecular nature of host-pathogen interactions and of pathogen- or vaccine-induced antibody responses. The first step of this approach is the three-dimensional structure determination of the antigen using structural biology tools such as X-ray crystallography, cryo-electron microscopy and NMR. Epitope mapping of an antigen that is recognized and bound by antibodies is key to vaccine development This will provide a comprehensive dataset at atomic level which is necessary to engineer new constructs with better properties in terms of elicitation of the antibody response, stability in solution and ease of production.
The aim of this re-engineering process is to obtain an antigen that is more effective in eliciting an optimal B cell response. Based on the analysis of antigen-antibody interaction network, the candidate vaccine antigen may contain only those residues essential to reproduce the protective epitopes. Molecular modeling is extremely helpful in designing optimal re-engineered antigens, through the identification of mutations that stabilize immunogenic conformations of epitopes. Moreover, residues outside the epitope can be mutated to improve antigen production yields. Another important issue in antigen re-engineering is the improvement of antigen thermal stability, through the identification of stability-enhancing mutations or through the insertion of the designed antigen in stable protein scaffolds. Eventually, the immunogenicity and efficacy of the candidate vaccine must be tested in animal models [21].
While glyco- or peptide-based subunit vaccines offer improved safety and more precise targeting, they usually require conjugation to immunogenic molecules (i.e., carrier proteins or immunogenic Th sequences), or presentation in multimeric format (virus-like particles or nanoparticles) to achieve optimal immune responses [22], which can be further enhanced by inclusion of an immunomodulatory adjuvant. Notably, issues of immunodominance are a critical aspect to designing optimized vaccines and need to be considered when selecting the epitope in order to elicit focused, protective broadly neutralizing antibody responses directed against naturally non-immunodominant conserved epitopes [23].
Another key element for rational vaccine design is the presentation of the antigen in its relevant native-like conformation, enabling optimal antibody recognition of the antigenic epitope within the quaternary protein structure. Several approaches have been developed to modulate these epitopes to favor conformationally relevant states of the antigen [24], including side chain cross-linking, grafting the epitope into a larger scaffold and other rationally designed chemical modifications leading to enhanced antibody binding affinity [25]. With the development of chemical technologies, such as covalent conjugation of immunogenic carrier proteins to polysaccharides, subunit vaccines have substantially advanced and have provided improved memory responses, thus addressing a key limitation suffered from saccharides vaccines. [26].
Several prototypes of structure-based vaccine development have been evaluated. One example represents the vaccine development against respiratory syncytial virus (RSV). In this case, specific epitopes belonging to the F glycoprotein (sites 0, III, and V) present potent neutralizing activity, 10 to 100 times greater than that observed for clinically used monoclonal antibody palivizumab (Synagis®). However, these key epitopes are well exposed only in the pre-fusion trimeric conformation of the RSV-F protein. Structural vaccinology was fundamental to generate stabilized pre-F trimers that preserved key epitopes in the proper conformation to elicit the most neutralizing activity in human sera [27][28]. Another example is the licensed 4-component vaccine against N. meningitidis serogroup B (MenB) composed of three recombinant proteins and a bacterial membrane vesicle selected using a reverse vaccinology approach [29]. Structure-based design was subsequently used to generate a vaccine made of chimeric antigens, by retaining epitopes from two antigens (fHbp and PorA), thus potentiating its power to elicit functional humoral immune responses against MenB [30]. These two examples illustrate that structural vaccinology can generate novel vaccine antigen candidates with improved characteristics for antibody-based strategies.
The potential improvement in the identification of antigenic determinants expressed on cancer cells enabled the design of therapeutic subunit vaccine to treat cancer [31][32][33]. Although the use of cancer vaccines awaits meaningful clinical benefits several tumor-associated carbohydrate antigens (TACAs) are currently largely validated and have been used to design promising molecular vaccines. Conjugation of these TACAs to proteins or the development of altered-self, i.e., more immunogenic, TACA analogues are examples of current strategies to make TACA-based vaccines able to break immune tolerance and elicit cancer antigen-specific antibodies [34].
2.2. Rational Antigens for Cell-Mediated Immunity
A considerable challenge in vaccinology is the design of vaccines against pathogens for which antibody immune responses are not protective [35]. To develop T cell vaccines, it is important to have an antigen that elicits potent cellular immune responses as well as an easy methodology to test the validation of the vaccine design in vivo and in vitro. In this regard, proteomics approaches with virulence factors of pathogens that induce potent CD4+ and CD8+ T cell responses are worthy methods to de-sign vaccines using reverse vaccinology approaches [36]. Using Listeria monocytogenes as a model pathogen Kono and co-workers and Calderon-Gonzalez and co-workers and DCs loaded with peptides of two virulence factors of this pathogen, listeriolysin O (LLO) and the glyceraldehyde-3-phosphate dehydrogenase (GAPDH) prepared vaccines that confer listeriosis protection [37][38]. Later, a methodology that combines bioinformatic analysis to screen for the best MHC binders, delayed type hypersensitivity (DTH) to test the best T cell mediated inducers and analysis of cytokines released by DC loaded with the epitopes and an adjuvant to search for epitopes inducing only Th1 and not Th2 cytokines [39] (Figure 2). Consequently, MHC binding epitopes and inducers of Th1 cytokines were selected and included in the peptide sequences contributing to immune protection. DCs loaded with those epitopes that meet the highest MHC binding, T cell induction and high levels of Th1 cytokines were validated as efficient epitopes for vaccination against L. monocytogenes challenge. This approach helps to predict other epitopes for vaccines against listeriosis and other pathogens as mycobacteria and streptococci [40]. Other proteomics approaches similar to this one also helped to design.
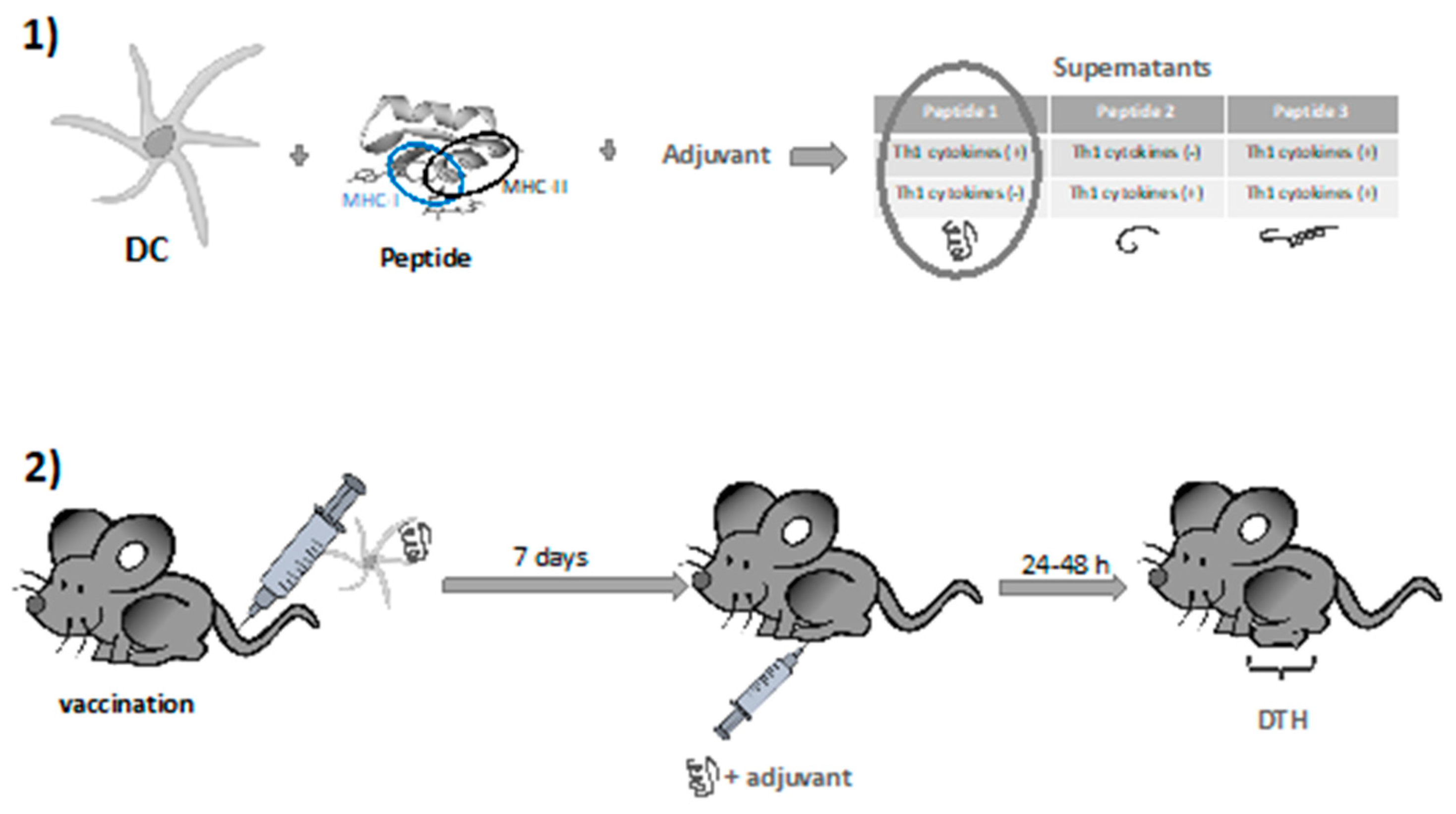
Figure 2. Methodology using dendritic cells (DCs) and adjuvants to select vaccine antigens for cell- mediated immunity. (1) DC loaded with peptides, good Major histocompatibility (MHC)-I and MHC-II binders and a Th1 adjuvant are checked for Th1 and Th2 cytokine releases. (2) Peptides inducing Th1 cytokines are selected and validated in mice vaccinated with DCs loaded with the peptides and inoculated into the footpads to measured delayed hypersensitivity (DTH) reactions. Peptides inducing strong DTH reactions are considered good candidates for future vaccines.
3. Choice of Immunomodulation
3.1. Rational Vaccine Adjuvant Design
For the past few decades, the focus of new vaccine development has been on the antigen(s) and new ways to present antigen. Optimization of the immune response with the use of different types of adjuvant has received less attention. Typically, when preliminary studies with a “standard” adjuvant does not show the desired (protective) response, the researchers will seek another type of antigen, instead of searching for an appropriate adjuvant that may provide the suitable or preferred immune response. Often the desired adjuvant or formulation is unknown or not available. Nevertheless, there have been noteworthy achievements in improving existing vaccines by introducing specific adjuvants as well as new vaccine delivery methods [41][42].
Live attenuated vaccines, however, do not usually require adjuvants since the resulting immune response is a result from the attenuated microbe and its attempt to multiply. However, adjuvants come in many forms [43] and may have many functions as shown in Table 1.
Table 1. Pharmaceutical and Immunological Functions of vaccine adjuvants.
| Pharmaceutical function (affecting vaccine antigen delivery) |
| Prolong antigen residence time at the site of administration |
| Protect the nature of the antigen |
| Prevent the antigen from degradation (improve stability) |
| Protect the 3D structure of the exposed antigen epitope(s) |
| Induce an environment that mimics the infection |
| Secure absorption into the lymphatics over systemic circulation |
| Decrease the number of boosters required for successful immune response |
| Have a good safety and toxicological profile |
| Immunological function (impact on immune function) |
| Attract antigen presenting cells to the site of administration |
| Augment immune response type: e.g., Th1, Th2, Th3 or Th17 |
| Augment the generation of memory cells |
| Augment mucosal, or systemic responses |
| Improve the generation of neutralizing antibodies and /or effector T cells |
3.2. Rationally Designed Vaccine Adjuvants
Molecularly defined subunit vaccines are generally safer but often lack immunogenicity, due to the absence of key additional structural components needed for efficient activation of innate immunity. Such vaccines require the coadministration of an adjuvant [44]. Few adjuvants are already licensed as part of vaccine formulations and some others are being used in the clinic [45]. However, the development of adjuvants has been traditionally an empirical process [46] as their molecular mechanisms of adjuvant activity are not fully understood [43][47][48], hindering the rational design of improved, less toxic adjuvants for optimal matching with selected vaccine antigens [49]. For long the immunological function of vaccine adjuvants has been subject of speculation [43].
It is not straightforward to translate the newly discovered mechanisms of adjuvant activity to generally applicable approaches for rationally designed vaccines. Thus, a thorough knowledge of adjuvants and their immunological effects is needed to realize the full potential of rational vaccine design. [50]. These critical vaccine elements come in many forms and serve to initiate, accelerate, amplify, improve and prolong the immunological responses to antigens. Vaccine adjuvants have been classified in vaccine antigen delivery systems (facilitating immune signal 1) or director immunostimulatory molecules (facilitation immune signal 2) or a combination of both [43].
The most common and long-time used immunoadjuvant alum has been extensively reviewed before [48][49], but mostly induces improved antibody responses. Other more recent clinical stage adjuvants include oil-based emulsions, such as MF-59, saponins, ISCOMs, oxoadenines, C-type lectin ligands. One prominent category includes Toll-like receptor agonists such as poly(I:C), the family of imidazoquinoline adjuvants (resiquimod, imiquimod), GLA, CpG motifs or flagellin. Another adjuvant group includes particle adjuvants, mimicking micro-organism structures, including nano- or microparticles or virus-mimicking polymers, such as polyphosphazene polyplexes PCPP and PCEP. Modularly composed AS0 adjuvants of Glaxo Smith Kline (GSK) and the CAF adjuvants of the Statens Serum Institute, have been extensively reviewed before [51][52]. All these systems will not be described in this review. Instead, a limited number of prototype examples of rationally designed vaccine adjuvants are described below.
3.2.1. Natural Lipid A as Source of Inspiration for New Adjuvants
The lipid A molecule is the immunostimulant component of Gram-negative bacterial lipopolysaccharide (LPS). It consists of a β-(1→6)-linked diglucosamine backbone with different patterns of acylation and phosphorylation [53]. The lipid A structure activates the TLR4/MD-2 immune receptor complex that mediates gene expression and secretion of pro-inflammatory cytokines. Through activation and regulation of various dendritic cell functions TLR4 activation bridges innate and adaptive immunity by inducing signals that are vitally involved in the initiation of adaptive immune responses [54]. TLR4 activation by lipid A is structure dependent. The bis-phosphorylated hexa-acylated lipid A species with a 4/2 distribution of the acyl chains (E. coli type) is the most toxic and its recognition and binding by the TLR4/MD-2 binary complex can lead to sepsis. Changing the number and length of the acyl chains or the phosphate decorations decreases its recognition by the immune system receptor [55][56]. A vast diversity of lipid A molecules can be found as moieties of Gram-negative bacterial LPS. Acyl chains can be absent (de-acylation) or hydroxylated [57] or more acyl chains can be added such as palmitate limiting immune system recognition [58]. The removal of the anomeric phosphate group of the E. coli type lipid A makes the molecule only moderately active. The phosphate groups can be absent or replaced by monosaccharides [59][60][61][62] or can also be decorated by phosphoethanolamine or cationic monosaccharides as 4-amino-4-deoxy-β-l-arabinose to neutralize their negative charge altering immune recognition [63][64]. In addition, in nature, the glucosamine disaccharide can be replaced by the 2,3-diamino-2,3-dideoxy-d-Glc disaccharide [65][66]. There is a vast number of lipid A molecules that are partial agonists and antagonists of TLR4. Particularly, gut microbiota have proven to be a tremendous library of lipid A molecules with different activities [67]. Lipid A is also capable of activating C-type lectin receptors (CTLR) [68], and caspases [69][70][71][72]. In addition, some atypical LPSs can activate TLR-2, although the chemical determinants responsible of this interaction are not yet clear [61][73].
For the structural reasons explained above, some natural LPS could be used as vaccine adjuvants. For example, Brucella abortus LPS showed promising results in various formulations [74][75], as well as the naturally occurring monophosphorylated lipid A of Bacteroides thetaiotaomicron and Prevotella intermedia [76]. There are also LPS-modified adjuvants like the O-deacylated lipooligosaccharide from E. coli J5 [77] or the broadly used adjuvant Mono-Phosphoryl Lipid A (MPL) (Figure 3) [78]. MPL is derived by the removal of the anomeric phosphate group of the hepta-acylated lipid A of S. minnesota R595, which reduces significantly the TLR4-driven activity and toxicity. Decreased TLR4 activation induced by MPL derives from a less efficient dimerization of TLR4/MD-2/MPL complexes due to the absence of the phosphate group which the weakens proteins’ interaction at the dimerization interface [79][80][81][82]. MPL formulations are incorporated in approved vaccine preparations; moreover, MPL has also been used as a starting point of further modifications to develop new adjuvants [83][84].
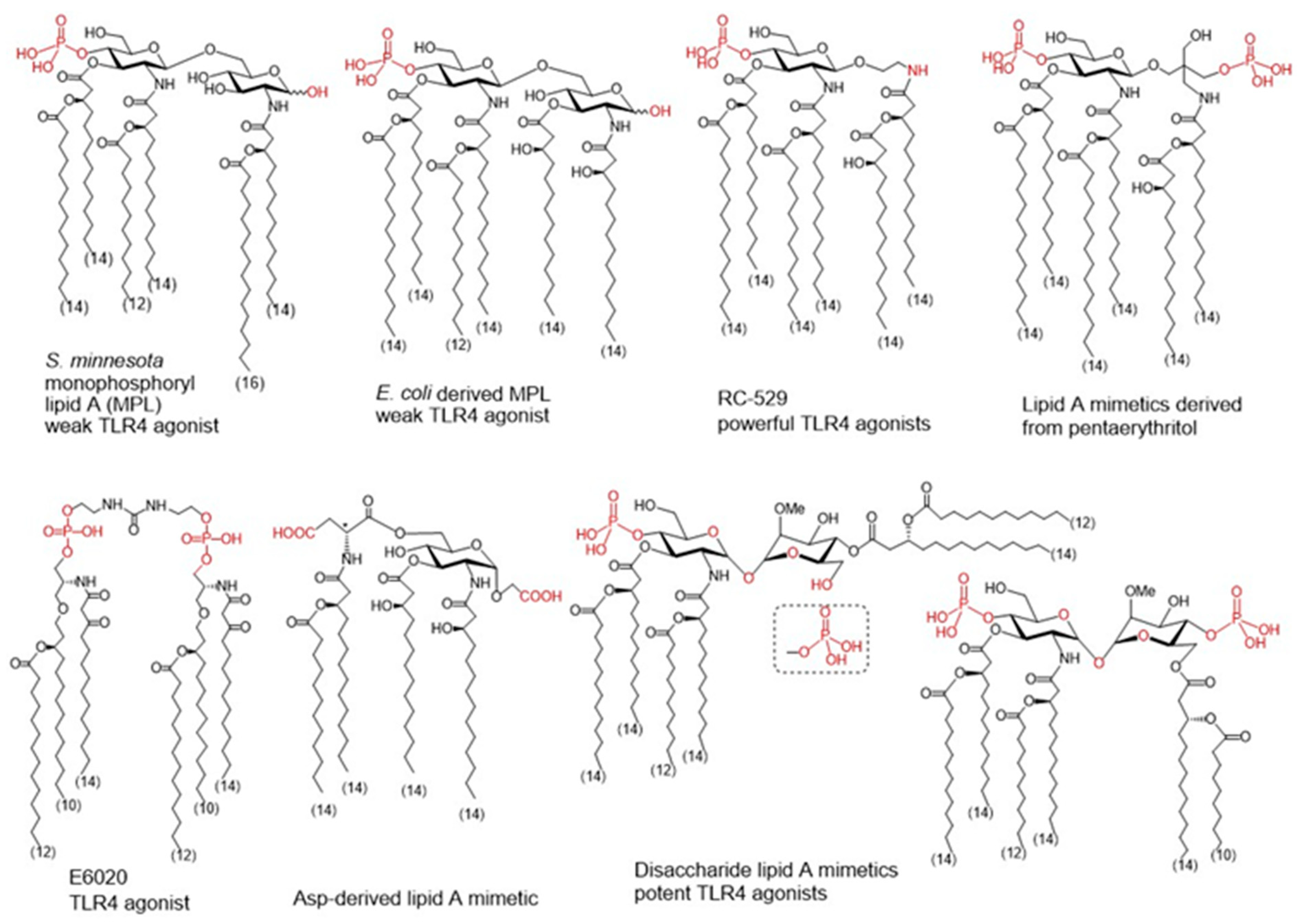
Figure 3. Monophpsphoryl lipids (MPLs)Ls and synthetic lipid A mimetics as promising vaccine adjuvant candidates.
To improve specific adjuvant properties and to modify or enhance immune stimulating activity, different synthetic lipid A analogues and lipid A mimetics have been prepared as vaccine adjuvant candidates. Chemical synthesis provides access to structurally defined molecules free of any biological contaminations and allows tremendous improvements in structure-activity relationships studies. Basically, all synthetic lipid A mimetics mirrored the basic architecture of their parent lipid A and were composed of a hydrophilic polar/charged head group and a hydrophobic lipid region. Simplification of the lipid A structure by replacing one or both glucosamine (GlcN) rings for linear or branched aglycons. Thus, GSK Biologicals developed aminoalkyl glucosaminide phosphates (AGPs) where the proximal GlcN ring of lipid A was omitted furnishing in this way a polar head group with rationalized structure [85]. Variable β-hydroxyacyl and alkyl chains were chemically attached to the polar head group and selected AGPs reached pre-clinical/clinical development which highlighted RC-529 as a potent vaccine adjuvant (Figure 3) [86]. Lipid A mimetics containing pentaerythritol in place of proximal GlcN residue were developed by Biomira Inc. (Edmonton, AB, Canada) as potent cytokine secreting agonists. A pentaerythritol-derived lipid A mimetic demonstrated adjuvant properties through enhancement of antigen-specific T cell activation in a synthetic liposomal vaccine system [87]. Replacement of the distal GlcN moiety of lipid A for the acidic amino acid Asp and the glycosidic phosphate group for a carboxyl group (Asp-derived lipid A mimetic) ensured immunostimulating potential despite of only four lipid chains attached [88]. Further structure simplification led to development of an acyclic lipid A mimetic E6020 (Eisai, Tokyo, Japan) consisting of a hexaacylated flexible linear backbone (Figure 3) [89]. The antibody response to vaccines co-administered with this TLR4 agonist E6020 led to a mixed Th1/Th2 response [90]. In addition, newly developed disaccharide lipid A mimetics profited from a conformational rigidity of their nonreducing disaccharide backbone and exhibited picomolar affinity for TLR4 [91]. Thus, adjustable TLR4 activation and graded induction of cellular pro-inflammatory responses renders these glycolipids promising vaccine adjuvant candidates [92].
3.2.2. Rational Design of Allostatine Adjuvant
Alloferons are a group of natural peptides isolated from insects that can stimulate human natural killer (NK)cell cytotoxicity towards cancer cells. These peptides are originally isolated from the hemolymph of maggots from the blowfly Calliphora vicina. A striking feature of larval hemolymph is that the hemocytes possess a cytotoxic activity functionally analogous to human NK cells. When stimulated by bacteria, these larvae produce high levels of potent defensive molecules typical of the insect immune system which accumulate in the hemolymph [93]. These include Alloferon1 and Alloferon2, two virtually indistinguishable peptides consisting of 13 and 12 amino acids respectively. Both Alloferons stimulate in vitro natural cytotoxicity of human blood mononuclear cells. Alloferon1 stimulates natural cytotoxicity in human in vitro models as well as antiviral and anticancer activities in mouse models in vivo. In a search for a molecule with higher antitumoral activity and cancer immunotherapy potential, the primary structure of Alloferon1 has been modified to obtain the peptide Allostatine, by changing two amino acids. The substitution of two amino acids in the Alloferon sequence was designed to simulate a pattern typical of the human immunoglobulin [94] belonging to the immunoglobulin heavy chain CDR3 region which is very well conserved between mammalian genomes, notwithstanding that CDR3 region is the most variable section of immunoglobulins. The molecular mechanism of action of Allostatine is still not clear. In silico simulations suggested NKG2D as a possible target for this peptide, while in vitro experiments showed that low, ng/mL concentrations of Allostatine in culture medium cause rearrangement of NK and T cells receptors, stimulating NK cells cytotoxic activity against cancer cells increasing the number of IFN-gamma and IL2 producing cells [95]. Allostatine manifested strong adjuvant properties in a mouse P388/DBA2 tumor transplantation model when combined with a vaccine consisting of X-ray inactivated tumor cells. While the vaccine alone demonstrated only a weak tumoristatic effect in about 25% of recipients, The vaccine in combination with Allostatine caused a tumoristatic effect in approximately 65% of recipients and prevented tumor occurrence in another 30% (resulting in a positive influence on 95% of recipients) [94]. Hence, the designed peptide Allostatine therefore, possesses characteristics of an adjuvant boosting cancer therapeutic vaccines efficacy.
3.2.3. Nod2 Agonists as Rationally Designed Vaccine Adjuvants
Nucleotide-binding oligomerization domain-containing protein 2 (NOD2) is a cytoplasmic pattern recognition receptor involved in both innate as well as adaptive immune responses and therefore constitutes an excellent target for rationally designed vaccine adjuvant ligands [96]. Muramyl dipeptide (MDP) is the smallest structural subunit of bacterial peptidoglycan capable of eliciting NOD2 activation, which leads to pro-inflammatory and antimicrobial responses characterized by the secretion of cytokines, induction of autophagy and production of antimicrobial peptides [96]. Activation of NOD2 itself is sufficient to shape the adaptive immune response towards a Th2 response [97]. Incidentally, NOD2 agonistic activities have been shown to strongly correlate with their adjuvant properties [98]. NOD2 agonists also amplify the adjuvant potential of TLR ligands and alter the magnitude, persistence and the type of response towards the Th1 type [99]. Interestingly, engagement of NOD2 proved to be essential for antigen-specific mucosal and systemic responses of mucosal vaccines [100][101]. This is noteworthy in light of the fact that the majority of pathogens gain entry through mucosal sites and given the shortage of mucosal adjuvants, the use of NOD2 agonists in cancer vaccines was also highlighted [102].
Although MDP (Figure 4 (1)) is predominantly responsible for the efficacy of Freund’s complete adjuvant, it suffers from pyrogenicity and rapid elimination as a single administrated molecule [99]. To that end, many chemically modified derivatives of the parent MDP molecule (Figure 4) have been synthesized with the aim of reducing its toxicity and improve its pharmacokinetic properties. Of the hydrophilic derivatives known to mainly induce a Th2-type response, murabutide (Figure 4 (2) and temurtide (Figure 4 (3) emerged as the most interesting candidates for further development as vaccine adjuvants, while muramyl tripeptide phosphatidylethanolamine (mifamurtide; MTP-PE) (Figure 4 (4), B30-MDP (Figure 4 (5) and romurtide (MDP-Lys (L18) (Figure 4(6) were the most prominent lipophilic derivatives, which tend to augment the Th1-type immune reaction [102].
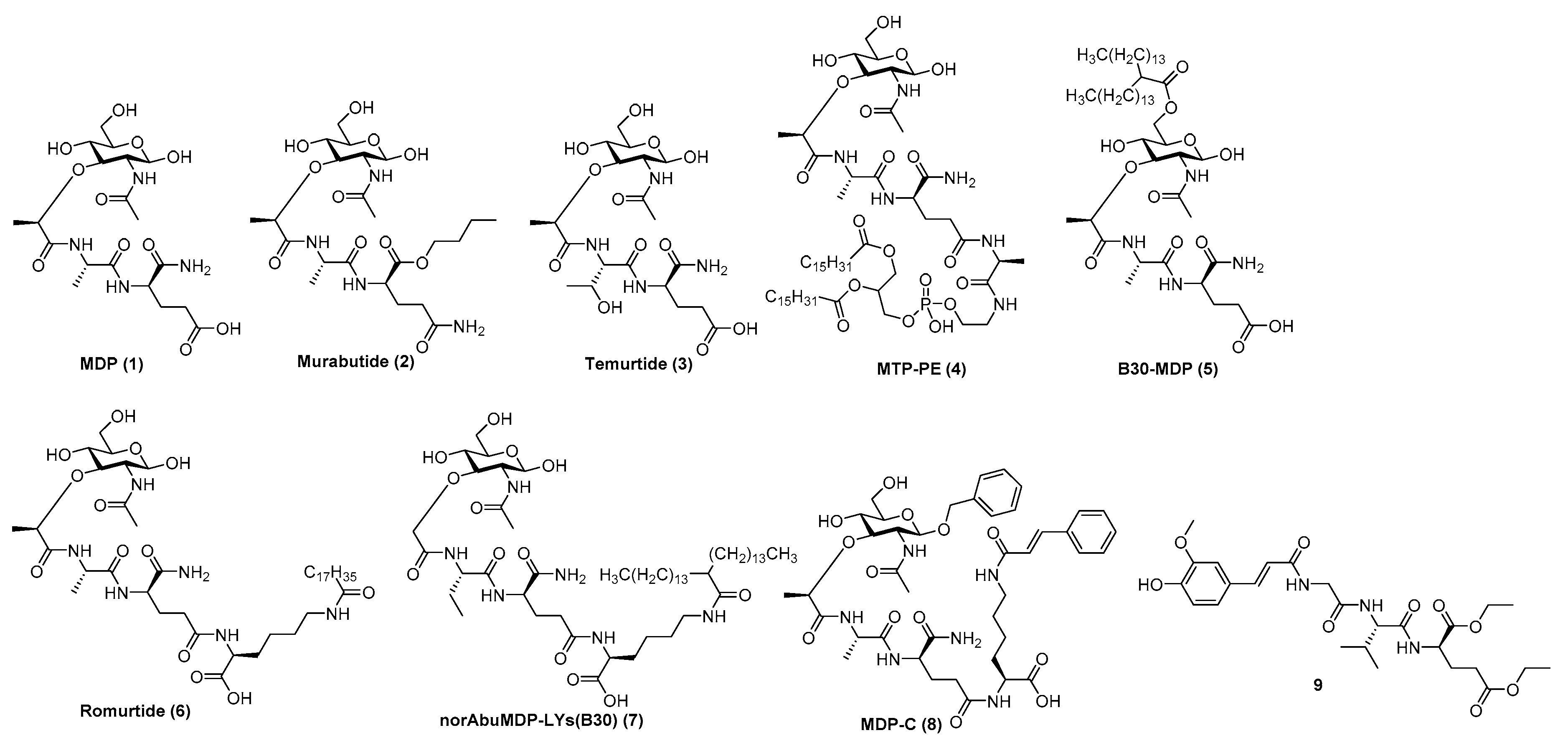
Figure 4. MDP (1), its most prominent pharmacologically active derivatives (2–8) and a desmuramylpeptide derivative with NOD2 activity (9).
Substitution of the N-acetyl group by an N-glycolyl group produces N-glycolyl-MDP, a distinctive feature of the BCG vaccine, which exhibits superior adjuvant activity. The l-Ala position is also available to other amino acids, since l-Ser, l-Val and l-Thr peptide analogs retained the adjuvant activity of the parent molecule [98]. The d-iGln moiety, on the other hand, is less amenable to substitution; it can only be replaced by either d-Gln or d-Glu (including their esterified forms) as exemplified by murabutide or muradimetide [96]. Murabutide is apyrogenic and well tolerated by humans [103], therefore its adjuvant effect was assessed following administration with the fluid phase of tetanus toxoid vaccine, in which case significantly higher IgG levels to toxoid were found in the group receiving vaccine with murabutide compared to the group given the vaccine alone [104]. Its adjuvant capacities were further underlined using a combination of murabutide and a synthetic hepatitis B antigen with increased levels of antigen-specific antibodies [105]. Recently, Jackson et al. reported on the ability of murabutide to induce a robust and durable IgG and IgA antibody response to Norwalk virus following intranasal vaccination which proved its ability to act as a potent mucosal adjuvant [101]. Temurtide is a threonine-based MDP derivative, used as an active principle of the SAF-1 formulation, which has been tested as adjuvant in preclinical trials in guinea pigs and mice. It successfully increased the formation of IgG2a antibodies against HBsAg [106].
The introduction of lipophilic groups into the structures of NOD2 agonists has been shown to strongly enhance the cellular immune response and overall increased the immunostimulatory adjuvant activity of the compounds. MTP-PE (Figure 4(4) was evaluated in Phase I clinical trials as a vaccine adjuvant in human immunodeficiency virus type I vaccines, but failed to improve their immunogenicity, while causing increased reactogenicity [107]. Decoration of the muramyl residue 6-OH group with a lipophilic linear/branched fatty acid structural feature resulted in derivatives with noteworthy adjuvant properties, as exemplified by B30-MDP [96]. Further structural optimization, which entailed the extension of the peptide stem with N-stearoyl-l-Lys, led to the discovery of MDP-Lys(L18) (also known as romurtide or muroctasine). A combination of MDP-Lys(L18) and B30-MDP has shown promising results in mice, which produced higher antibody titres against rHBsAg after intraperitoneal injection [108] and increased the humoral and cellular response against an inactivated hantavirus vaccine [109]. A close structural analog norAbuMDP-Lys(B30) (Figure 4 (7), which proved effective as an adjuvant for Borrelia burgdorferi antigen rOspA, also features a B30-acylated Lys-extension [110]. Similarly, MDP-C (Figure 4 (8) carries a N-cinnamoyl-l-Lys moiety and showed promising results in a mouse model by increasing the levels of anti-HBs antibodies [111]. Desmuramylpeptides are MDP derivatives in which the sugar N-acetylmuramyl moiety (MurNAc) is replaced by a hydrophobic group. The Trans-feruloyl moiety has recently been identified as an excellent MurNAc mimetic, resulting in a low nanomolar NOD2 agonist (Figure 4 (9), which induced ovalbumin-specific IgG titers in a mouse model of adjuvancy [112].
3.2.4. QS-21-Based Synthetic Saponin Adjuvants
A number of adjuvant analogues have been developed based on natural product adjuvants leveraging detailed structure–activity relationship studies, such as synthetic saponin variants derived from QS-21 (Figure 5a) and the Quillaja saponin (QS) family [113].
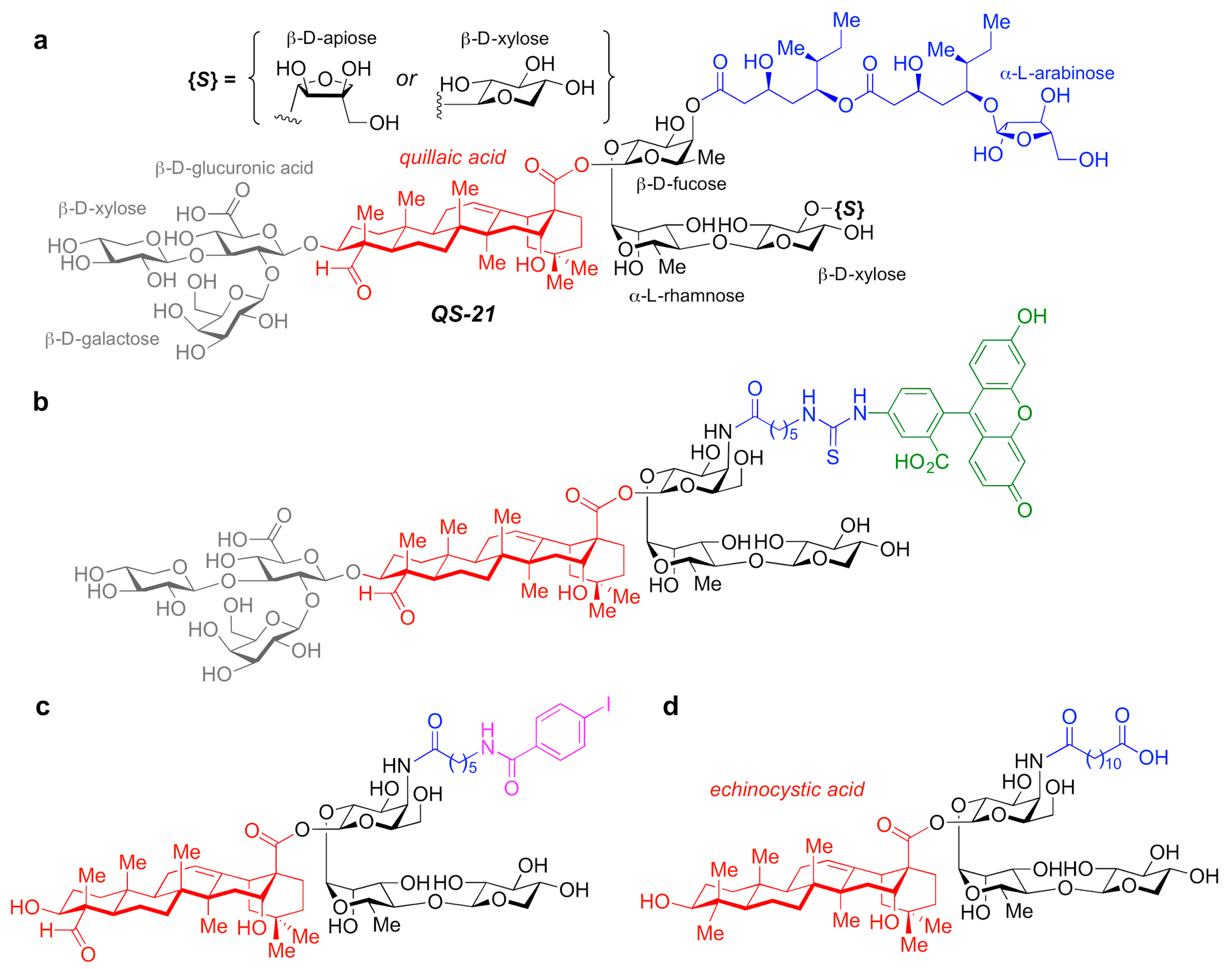
Figure 5. (a) Chemical structure of QS-21 saponin natural product adjuvant. (b–d) Synthetic saponin variants based on QS-21.
In particular, QS-21 is a saponin natural product with a long history and great potential as an adjuvant. It elicits both antibody and cellular immune responses, including cytotoxic T lymphocytes, and has been recently approved in combination with MPLA as part of the AS01 adjuvant system in vaccines against malaria and shingles [114]. However, despite its promise, QS-21 suffers from several drawbacks, including scarcity, heterogeneity, chemical instability and dose-limiting toxicity, which have hampered its more widespread use in human vaccines. As such, the discovery of new, improved QS-21 variants has been at the forefront. In this context, Fernández-Tejada et al. identified key structural features of QS-21 that are important for adjuvant activity [115][116] and have developed a variety of simplified, synthetically accessible saponin derivatives that induce antibody responses comparable to QS-21 with drastically reduced toxicity (Figure 5b–d) [117][118]. Moreover, these structurally simpler saponin scaffolds were leveraged for the development of saponin variants bearing fluorescent and radioiodinated tags (Figure 5b,c). These saponin probes were exploited in imaging and biodistribution studies that revealed internalization of active variants into dendritic cells and accumulation in the lymph nodes, which suggests a role for adjuvant-active QS variants in the trafficking of antigens by APCs to the draining lymph nodes [117].
Additional studies on the detailed immunological profile of these saponins are warranted and in progress, with the aim to elucidate the precise functional and molecular roles of these adjuvants, also in the context of adjuvant systems and anti-cancer vaccines.
3.3. The VLP-Based Vaccine Platform and CpG ODNs as Immunoprotective Vaccine Adjuvants
The spread of COVID-19 highlighted the need for swift vaccine development in a global pandemic and over 320 vaccine trials were conducted worldwide at time of writing [119][120]. Historically live attenuated viral vaccines could be selected as one of the most effective and protective platforms during a pandemic. However, the challenge involved in rapid generation of an attenuated SARS-CoV-2, as well as recent advances in molecular biology and immunology, promoted the evaluation of faster alternative strategies, such as Virus Like Particles (VLPs), as a convenient and effective approaches against controlling COVID-19 pandemic.
VLPs are macromolecular self-assembling structures that closely resemble the native forms of viruses. One of the superior features of these “stunt viruses” is that they are non-infectious since they lack viral genetic content. Therefore, VLPs are safer than whole-pathogen-based vaccines such as those containing attenuated viruses. VLPs can be developed through expression of individual viral structural proteins following transfection which can self-assemble into the VLPs before being released into the extracellular environment from the producer cells. The success and licensing of the multivalent VLP-based vaccines for human papilloma virus and Hepatitis B validated the safety and efficacy of the vaccine concept for VLPs [121][122], which supports the utilization of this platform to further develop effective vaccines against newly emerging infectious agents. A Coronovirus-like particle VLP vaccine is being evaluated in Phase 2/3 trials [123].
In the case of SARS-CoV2 for example, four structural proteins of SARS-CoV-2, namely, spike, envelope, membrane and nucleocapsid could be cloned within proper expression vector(s) and expressed in a suitable producer cell line that could range from mammalian to insect, to even yeast or plant cells. The immunogenicity and strength of protective capacity of these vaccine candidates could be further amplified by the use of a proper Th1 immunity-supporting biological adjuvants such as CpG oligodeoxynucleotides (CpG ODN hereafter) [124]. Development of effective vaccine mediated immune responses relies on the use of vaccine adjuvants capable of enhancing and directing the adaptive immune response to the antigen. When used as vaccine adjuvants, type I interferon inducing agents can elicit both potent effector/memory T cell responses and humoral immunity.
Specific sequences of single stranded synthetic oligodeoxynucleotides containing unmethylated cytosine-phosphate-guanine oligodeoxynucleotide motifs (CpG ODN) stimulate type I interferon production via TLR9. Based on their differential activation of immune cells, four major classes of synthetic CpG ODNs have been defined. The K class ODNs are potent B cell activators that stimulate TNF-α secretion not interferon-α (IFNα), while D, C-, and P-class ODNs induce variable amounts of IFNα secretion [125][126]. The D-class ODNs are the most potent IFNα inducers but form multimers which complicates their GMP manufacture. There have been only three clinical trials to date evaluating D-class ODNs as either a vaccine adjuvants and/or immunotherapeutic applications. All three studies harnessed a stabilized version of this ODN class following packaging into virus like particles consisting of the bacteriophage Qß coat protein. Interestingly, our recent studies confirmed that inclusion of CpG ODNs within SARS-CoV-2 VLPs elicited pronounced humoral and cell mediated immunity against COVID-19 infection (Gursel et al., 2021 unpublished data).
References
- Zhu, Y.; An, X.; Zhang, X.; Qiao, Y.; Zheng, T.; Li, X. STING: A master regulator in the cancer-immunity cycle. Mol. Cancer 2019, 18, 1–15.
- Pulendran, B.; Li, S.; Nakaya, H.I. Systems Vaccinology. Immunity 2010, 33, 516–529.
- Rappuoli, R. Bridging the knowledge gaps in vaccine design. Nat. Biotechnol. 2007, 25, 1361–1366.
- Brodin, P.; Jojic, V.; Gao, T.; Bhattacharya, S.; Angel, C.J.L.; Furman, D.; Shen-Orr, S.; Dekker, C.L.; Swan, G.E.; Butte, A.J.; et al. Variation in the Human Immune System Is Largely Driven by Non-Heritable Influences. Cell 2015, 160, 37–47.
- Cheung, P.; Vallania, F.; Warsinske, H.C.; Donato, M.; Schaffert, S.; Chang, S.E.; Dvorak, M.; Dekker, C.L.; Davis, M.M.; Utz, P.J.; et al. Single-Cell Chromatin Modification Profiling Reveals Increased Epigenetic Variations with Aging. Cell 2018, 173, 1385–1397.e14.
- Hagan, T.; Cortese, M.; Rouphael, N.; Boudreau, C.; Linde, C.; Maddur, M.S.; Das, J.; Wang, H.; Guthmiller, J.; Zheng, N.-Y.; et al. Antibiotics-Driven Gut Microbiome Perturbation Alters Immunity to Vaccines in Humans. Cell 2019, 178, 1313–1328.e13.
- Cortese, M.; Sherman, A.C.; Rouphael, N.G.; Pulendran, B. Systems Biological Analysis of Immune Response to Influenza Vaccination. Cold Spring Harb. Perspect. Med. 2020, a038596.
- Chaussabel, D.; Quinn, C.; Shen, J.; Patel, P.; Glaser, C.; Baldwin, N.; Stichweh, D.; Blankenship, D.; Li, L.; Munagala, I.; et al. A modu-lar analysis framework for blood genomics studies: Application to systemic lupus erythematosus. Immunity 2008, 29, 150–164.
- Li, S.; Rouphael, N.; Duraisingham, S.S.; Romero-Steiner, S.; Presnell, S.R.; Davis, C.; Schmidt, D.S.; E Johnson, S.; Milton, A.; Rajam, G.; et al. Molecular signatures of antibody responses derived from a systems biology study of five human vaccines. Nat. Immunol. 2014, 15, 195–204.
- Wimmers, F.; Pulendran, B. Emerging technologies for systems vaccinology—Multiomics integration and single-cell (epi)genomic profiling. Curr. Opin. Immunol. 2020, 65, 57–64.
- Li, S.; Sullivan, N.L.; Rouphael, N.; Yu, T.; Banton, S.; Maddur, M.S.; McCausland, M.; Chiu, C.; Canniff, J.; Dubey, S.; et al. Metabolic Phenotypes of Response to Vaccination in Humans. Cell 2017, 169, 862–877.e17.
- Braun, R.O.; Brunner, L.; Wyler, K.; Auray, G.; Garcia-Nicolas, O.; Python, S.; Zumkehr, B.; Gaschen, V.; Stoffel, M.H.; Collin, N.; et al. System immunology-based identification of blood transcrip-tional modules correlating to antibody responses in sheep. NPJ Vaccines 2018, 3, 41.
- Matthijs, A.M.F.; Auray, G.; Jakob, V.; Garcia-Nicolas, O.; Braun, R.O.; Keller, I.; Bruggman, R.; Devriendt, B.; Boyen, F.; Guzman, C.A.; et al. Systems Immu-nology Characterization of Novel Vaccine Formulations for Mycoplasma hyopneumoniae Bacterins. Front. Immunol. 2019, 10, 1087.
- Tsang, J.S.; Dobaño, C.; VanDamme, P.; Moncunill, G.; Marchant, A.; Ben Othman, R.; Sadarangani, M.; Koff, W.C.; Kollmann, T.R. Improving Vaccine-Induced Immunity: Can Baseline Predict Outcome? Trends Immunol. 2020, 41, 457–465.
- Delany, I.; Rappuoli, R.; Seilb, K.L. Vaccines, reverse vaccinology and bacterial pathogenesis. CSH Perspect. 2013, 3, a012476.
- Jefferies, J.M.C.; Macdonald, E.; Faust, S.N. Clarke SC, 13-valent pneumococcal conjugate vaccine (PCV13). Hum. Vaccines 2011, 7, 1012–1018.
- Rappuoli, R.; Bottomley, M.J.; D’Oro, U.; Finco, O.; De Gregorio, E. Reverse vaccinology 2.0: Human immunology instructs vac-cine antigen design. J. Exp. Med. 2016, 213, 469–481.
- Burton, D.R. What Are the Most Powerful Immunogen Design Vaccine Strategies? Reverse Vaccinology 2.0 Shows Great Promise. Cold Spring Harb. Perspect. Biol. 2017, 9, 030262.
- Correia, B.E.; Bates, J.T.; Loomis, R.J.; Baneyx, G.; Carrico, C.; Jardine, J.G.; Rupert, P.B.; Correnti, C.; Kalyuzhniy, O.; Vittal, V.; et al. Proof of principle for epitope-focused vaccine design. Nat. Cell Biol. 2014, 507, 201–206.
- Graham, B.S.; Gilman, M.S.A.; McLellan, J.S. Structure-Based Vaccine Antigen Design. Annu. Rev. Med. 2019, 70, 91–104.
- Anasir, M.A. Chit Laa Poh. Structural Vaccinology for Viral Vaccine Design. Front Microbiol. 2019, 10, 738.
- Malonis, R.J.; Lai, J.R.; Vergnolle, O. Peptide-Based Vaccines: Current Progress and Future Challenges. Chem. Rev. 2020, 120, 3210–3229.
- Alam, S.M.; Dennison, S.M.; Aussedat, B.; Vohra, Y.; Park, P.K.; Fernández-Tejada, A.; Stewart, S.; Jaeger, F.H.; Anasti, K.; Blinn, J.H.; et al. Recognition of synthetic glycopeptides by HIV-1 broadly neutralizing antibodies and their unmutated ancestors. Proc. Natl. Acad. Sci. USA 2013, 110, 18214–18219.
- Kanekiyo, M.; Ellis, D.; King, N.P. New Vaccine Design and Delivery Technologies. J. Infect. Dis. 2019, 219, S88–S96.
- Compañón, I.; Guerreiro, A.; Mangini, V.; Castro-López, J.; Escudero-Casao, M.; Avenoza, A.; Busto, J.H.; Castillón, S.; Jiménez-Barbero, J.; Asensio, J.L.; et al. Structure-based design of potent tumor-associated antigens: Modulation of peptide presentation by single-atom O/S or O/Se substitutions at the glycosidic linkage. J. Am. Chem. Soc. 2019, 141, 4063–4072.
- Lewis, G.K.; DeVico, A.L.; Gallo, R.C. Antibody persistence and T cell balance: Two key factors confronting HIV vaccine development. Proc. Natl. Acad. Sci. USA 2014, 111, 15614.
- Joyce, M.G.; Zhang, B.; Ou, L.; Chen, M.; Chuang, G.-Y.; Druz, A.; Kong, W.-P.; Lai, Y.-T.; Rundlet, E.J.; Tsybovsky, Y.; et al. Iterative structure-based improvement of a fusion-glycoprotein vaccine against RSV. Nat. Struct. Mol. Biol. 2016, 23, 811–820.
- Ngwuta, J.O.; Chen, M.; Modjarrad, K.; Joyce, M.G.; Kanekiyo, M.; Kumar, A.; Yassine, H.M.; Moin, S.M.; Killikelly, A.M.; Chuang, G.-Y.; et al. Prefusion F–specific antibodies determine the magnitude of RSV neutralizing activity in human sera. Sci. Transl. Med. 2015, 7, 309ra162.
- Pizza, M.; Scarlato, V.; Masignani, V.; Giuliani, M.M.; Arico, B.; Comanducci, M.; Jennings, G.T.; Baldi, L.; Bartolini, E.; Capec-chi, B.; et al. Identification of vaccine candidates against serogroup B meningococ-cus by whole-genome sequencing. Science 2000, 287, 1816–1820.
- Hollingshead, S.; Jongerius, I.; Exley, R.M.; Johnson, S.; Lea, S.M.; Tang, C.M. Structure-based design of chimeric antigens for multivalent protein vaccines. Nat. Commun. 2018, 9, 1–10.
- Geldmacher, A.; Freier, A.; Losch, F.O.; Walden, P. Therapeutic vaccination for cancer immunotherapy: Antigen selection and clinical response. Human Vaccines 2011, 7, 115.
- Butterfield, L.H. Lessons learned from cancer vaccine trials and target antigen choice. Cancer Immunol. Immunother. 2016, 65, 805–812.
- Xu, Z.; Kulp, D.W. Protein engineering and particulate display of B-cell epitopes to facilitate development of novel vaccines. Curr. Opin. Immunol. 2019, 59, 49.
- Cheever, M.A.; Allison, J.P.; Ferris, A.S.; Finn, O.J.; Hastings, B.M.; Hecht, T.T.; Mellman, I.; Prindiville, S.A.; Steinman, R.M.; Viner, J.L.; et al. The prioritization of cancer an-tigens: A national cancer institute pilot project for the acceleration of translational research Clin. Cancer Res 2009, 15, 5323.
- Radford, K.J.; Caminschi, I. New generation of dendritic cell vaccines. Hum. Vaccines Immunother. 2013, 9, 259–264.
- Srivastava, S.; Sharma, S.K.; Srivastava, V.; Kumar, A. Proteomic Exploration of Listeria monocytogenes for the Purpose of Vaccine Designing Using a Reverse Vaccinology Approach. Int. J. Pept. Res. Ther. 2021, 27, 779–799.
- Calderon-Gonzalez, R.; Tobes, R.; Pareja, E.; Frande-Cabanes, E.; Petrovsky, N.; Alvarez-Dominguez, C. Identification and characterisation of T cell epitopes for incorporation into dendritic cell-delivered Listeria vaccines. J. Immunol. Methods 2015, 424, 111–119.
- Kono, M.; Nakamura, Y.; Suda, T.; Uchijima, M.; Tsujimura, K.; Nagata, T.; Giermasz, A.S.; Kalinski, P.; Nakamura, H.; Chida, K. En-hancement of protective immunity against intracellular bacteria using type-1 polarized dendritic cell (DC) vaccine. Vaccine 2012, 30, 2633–2639.
- Calderon-Gonzalez, R.; Frande-Cabanes EBronchalo-Vicente, L.; Lecea-Cuello, M.J.; Bosch-Martinez, A.; Fanarraga, M.L.; Yañez-Diaz, S.; Carrasco-Marin, E.; Alvarez-Dominguez, C. Cellular vaccines in listeriosis: Role of the Listeria antigen GAPDH. Front. Cell. Infect. Microbiol. 2014, 4, 22.
- Alvarez-Dominguez, C.; Salcines-Cuevas, D.; Teran-Navarro, H.; Calderon-Gonzalez, R.; Tober, R.; Garcia, I.; Grijalvo, S.; Paradela, A.; Seoane, A.; Sangari, F.J.; et al. Epitopes for multivalent vaccines against Listeria, Mycobacterium and Streptococcus spp: A novel role for glyceraldehyde-3-phosphate dehydrogenase. Front. Cell. Infect. Microbiol. 2020, 10, 573348.
- Robbins, A. Progress towards vaccines we need and do not have. Lancet 1990, 335, 1436–1438.
- Brown, F.; Dougan GMoey, E.M. A short history of vaccination. In Vaccine Design; Brown, F., Dougan, G., Moey, E.M., Eds.; John Wiley and Sons: Chichester, UK, 1993; pp. 1–6.
- Schijns, V.E. Immunological concepts of vaccine adjuvant activity. Curr. Opin. Immunol. 2020, 12, 456–463.
- Christensen, D. Vaccine adjuvants: Why and how. Hum. Vaccines Immunother. 2016, 12, 2709–2711.
- Del Giudice, G.; Rappuoli, R.; Didierlaurent, A.M. Correlates of adjuvanticity: A review on adjuvants in licensed vaccines. Semin. Immunol. 2018, 39, 14–21.
- O’Hagan, D.T.; Fox, C.B. New generation adjuvants—From empiricism to rational design. Vaccine 2015, 33, B14–B20.
- Ribeiro, C.M.; Schijns, V.E. Immunology of Vaccine Adjuvants. In Methods in Molecular Biology; Springer International Publishing: New York, NY, USA, 2009; Volume 626, pp. 1–14.
- Shi, S.; Zhu, H.; Xia, X.; Liang, Z.; Ma, X.; Sun, B. Vaccine adjuvants: Understanding the structure and mechanism of adjuvanticity. Vaccine 2019, 37, 3167–3178.
- Bonam, S.R.; Partidos, C.D.; Halmuthur, S.K.M.; Muller, S. An overview of novel adjuvants designed for improving vaccine efficacy. Trends Pharmacol. Sci. 2017, 38, 771–793.
- Rueckert, C.; Guzmán, C.A. Vaccines: From empirical development to rational design. PLoS Pathog. 2012, 8, e1003001.
- Garçon, N.; Chomez, P.; Van Mechelen, M. GlaxoSmithKline Adjuvant Systems in vaccines: Concepts, achievements and perspectives. Expert Rev. Vaccines 2007, 6, 723–739.
- Pedersen, G.K.; Andersen, P.; Christensen, D. Immunocorrelates of CAF family adjuvants. Semin. Immunol. 2018, 39, 4–13.
- Molinaro, A.; Holst, O.; Di Lorenzo, F.; Callaghan, M.; Nurisso, A.; D’Errico, G.; Zamyatina, A.; Peri, F.; Berisio, R.; Jerala, R.; et al. Chemistry of Lipid A: At the Heart of Innate Immunity. Chem. Eur. J. 2015, 21, 500–519.
- Iwasaki, A.; Medzhitov, R. Toll-like receptor control of the adaptive immune responses. Nat. Immunol. 2004, 5, 987–995.
- Schromm, A.B.; Brandenburg, K.; Loppnow, H.; Moran, A.P.; Koch, M.H.J.; Rietschel, E.T.; Seydel, U. Biological activities of lipopolysaccharides are determined by the shape of their lipid A portion. FEBS J. 2000, 267, 2008–2013.
- Kong, Q.; Six, D.A.; Liu, Q.; Gu, L.; Wang, S.; Alamuri, P.; Raetz, C.R.H.; Curtiss, R. Phosphate groups of lipid A are essential for Salmonella enterica serovar Typhimurium virulence and affect innate and adaptive immunity. Infect. Immun. 2012, 80, 3215–3224.
- Bhat, U.R.; Forsberg, L.S.; Carlson, R.W. Structure of lipid A component of Rhizobium leguminosarum bv. phaseoli lipopolysaccharide. Unique nonphosphorylated lipid A containing 2-amino- 2-deoxygluconate, galacturonate, and glu-cosamine. J. Biol. Chem. 1994, 269, 14402–14410.
- Raetz, C.R.H.; Reynolds, C.M.; Trent, M.S.; Bishop, R.E. Lipid A modification systems in Gram-negative bacteria. Annu. Rev. Biochem 2007, 76, 295–329.
- Plotz, B.M.; Lindner, B.; Stetter, K.O.; Holst, O. Characterization of a Novel Lipid A Containing D-Galacturonic Acid That Replaces Phosphate Residues. J. Biol. Chem. 2000, 275, 11222–11228.
- Silipo, A.; Vitiello, G.; Gully, D.; Sturiale, L.; Chaintreuil, C.; Fardoux, J.; Gargani, D.; Lee, H.I.; Kulkarni, G.; Busset, N.; et al. Covalently linked hopanoid-lipid A improves outer-membrane resistance of a Bradyrhizobium symbiont of legumes. Nat. Commun. 2014, 5, 5106.
- Di Lorenzo, F.; Pither, M.D.; Martufi, M.; Scarinci, I.; Guzmán-Caldentey, J.; Łakomiec, E.; Jachymek, W.; Bruijns, S.C.M.; Santamaría, S.M.; Frick, J.-S.; et al. Pairing Bacteroides vulgatus LPS Structure with Its Immunomodulatory Effects on Human Cellular Models. ACS Cent. Sci. 2020, 6, 1602–1616.
- Schwudke, D.; Linscheid, M.; Strauch, E.; Appel, B.; Zahringer, U.; Moll, H.; Muller, M.; Brecker, L.; Gronow, S.; Lindner, B. The Obligate Predatory Bdellovibrio bacteriovorus Possesses a Neutral Lipid A Containing α-D-Mannoses That Replace Phosphate Residues. J. Biol. Chem. 2003, 278, 27502–27512.
- Zhou, Z.; Ribeiro, A.A.; Lin, S.; Cotter, R.J.; Miller, S.I.; Raetz, C.R.H. Lipid A Modifications in Polymyxin-resistant Salmonella typhimurium. PRMA dependent 4-amino-4-deoxy-L-arabinose, and phosphoethanolamine incorporation. J. Biol. Chem. 2001, 276, 43111–43121.
- Silipo, A.; Molinaro, A.; Cescutti, P.; Bedini, E.; Rizzo, R.; Parrilli, M.; Lanzetta, R. Complete structural characterization of the lipid A fraction of a clinical strain of B. cepacia genomovar I lipopolysaccharide. Glycobiology 2005, 15, 561–570.
- Casabuono, A.C.; Czibener, C.; del Giudice, M.G.; Valguarnera, E.; Ugalde, J.E.; Couto, A.S. New Features in the Lipid A Structure of Brucella suis and Brucella abortus Lipopolysaccharide. J. Am. Chem. Soc. Mass. Spec. 2017, 28, 2716–2723.
- Zahringer, U.; Lindner, B.; Knirel, Y.A.; van den Akker, W.M.R.; Hiestand, R.; Heine, H.; Dehio, C. Structure and Biological Activity of the Short-chain Lipopolysaccharide from Bartonella henselae ATCC 49882T. J. Biol. Chem. 2004, 279, 21046–21054.
- Di Lorenzo, F.; de Castro, C.; Silipo, A.; Molinaro, A. Lipopolysaccharide structures of Gram-negative populations in the gut microbiota and effects on host interactions. FEMS Microbiol. Rev. 2019, 43, 257–272.
- Van Vliet, S.J.; Steeghs, L.; Bruijns, S.C.M.; Vaezirad, M.M.; Blok, C.S.; Busto, J.A.A.; Deken, M.; van Putten, J.P.M.; van Kooyk, Y. Variation of Neisseria gonorrhoeae Lipooligosaccharide Directs Dendritic Cell–Induced T Helper Responses. PLoS Pathog. 2009, 5, e1000625.
- Hagar, J.A.; Powell, D.A.; Aachoui, Y.; Ernst, R.K.; Miao, E.A. Cytoplasmic LPS activates caspase-11: Implications in TLR4-independent endotoxic shock. Science 2013, 341, 1250–1253.
- Kayagaki, N.; Wong, M.T.; Stowe, I.B.; Ramani, S.R.; Gonzalez, L.C.; Akashi-Takamura, S.; Miyake, K.; Zhang, J.; Lee, W.P.; Muszýnski, A.; et al. Noncanonical inflammasome activation by intracellular LPS in-dependent of TLR4. Science 2013, 341, 1246–1249.
- Shi, J.; Zhao, Y.; Wang, Y.; Gao, W.; Ding, J.; Li, P.; Hu, L.; Shao, F. Inflammatory caspases are innate immune receptors for intracellular LPS. Nature 2014, 514, 187–192.
- Viganò, E.; Diamond, C.E.; Spreafico, R.; Balachander, A.; Sobota, R.M.; Mortellaro, A. Human caspase-4 and caspase-5 regulate the one-step non-canonical inflammasome activation in monocytes. Nat. Commun. 2015, 6, 8761.
- Chavarría-Velázquez, C.O.; Torres-Martínez, A.C.; Montaño, L.F.; Rendón-Huerta, E.P. TLR2 activation induced by H. pylori LPS promotes the differential expression of claudin-4, -6, -7 and -9 via either STAT3 and ERK1/2 in AGS cells. Immunobiology 2018, 223, 38–48.
- Jamalan, M.; Ardestani, S.K.; Zeinali, M.; Mosaveri, N.; Taheri, M.M. Effectiveness of Brucella abortus lipopol-ysaccharide as an adjuvant for tuberculin PPD. Biologicals 2011, 39, 23–28.
- Kianmehr, Z.; Soleimanjahi, H.; Ardestani, S.K.; Fotouhi, F.; Abdoli, A. Influence of Brucella abortus lipopolysaccharide as an adjuvant on the immunogenicity of HPV-16 L1VLP vaccine in mice. Med. Microbiol. Immunol. 2015, 204, 205–213.
- Chilton, P.M.; Hadel, D.M.; To, T.T.; Mitchell, T.C.; Darveau, R.P. Adjuvant activity of naturally occurring monophos-phoryl lipopolysaccharide preparations from mucosa-associated bacteria. Infect. Immun. 2013, 81, 3317–3325.
- Ko, A.; Wui, S.R.; Ryu, J.I.; Do, H.T.T.; Lee, Y.J.; Lim, S.J.; Rhee, I.; Jung, D.I.; Park, J.-A.; Choi, J.-A.; et al. Comparison of the adjuvanticity of two adjuvant formulations containing de-O-acylated lipooligosaccharide on Japanese encephalitis vaccine in mice. Arch. Pharmacal Res. 2018, 41, 219–228.
- Casella, C.; Mitchell, T. Putting endotoxin to work for us: Monophosphoryl lipid A as a safe and effective vaccine adjuvant. Cell. Mol. Life Sci. 2008, 65, 3231–3240.
- Embry, C.A.; Franchi, L.; Nunez, G.; Mitchell, T.C. Mechanism of Impaired NLRP3 Inflammasome Priming by Mono-phosphoryl Lipid A. Sci. Signal. 2011, 4, ra28.
- Casella, C.R.; Mitchell, T.C. Inefficient TLR4/MD-2 Heterotetramerization by Monophosphoryl Lipid A. PLoS ONE 2013, 8, e62622.
- Tanimura, N.; Saitoh, S.-I.; Ohto, U.; Akashi-Takamura, S.; Fujimoto, Y.; Fukase, K.; Shimizu, T.; Miyake, K. The attenuated inflammation of MPL is due to the lack of CD14-dependent tight dimerization of the TLR4/MD2 complex at the plasma membrane. Int. Immunol. 2014, 26, 307–314.
- Pantel, A.; Cheong, C.; Dandamudi, D.; Shrestha, E.; Mehandru, S.; Brane, L.; Ruane, D.; Teixeira, A.; Bozzacco, L.; Steinman, R.M.; et al. A new synthetic TLR4 agonist, GLA, allows dendritic cells targeted with antigen to elicit Th1 T cell immunity in vivo. Eur. J. Immunol. 2012, 42, 101–109.
- Carter, D.; Fox, C.B.; Day, T.A.; Guderian, J.A.; Liang, H.; Rolf, T.; Vergara, J.; Sagawa, Z.K.; Ireton, G.; Orr, M.T.; et al. A structure-function approach to optimizing TLR4 ligands for human vaccines. Clin. Transl. Immunol. 2016, 5, e108.
- Gregg, K.A.; Harberts, E.; Gardner, F.M.; Pelletier, M.R.; Cayatte, C.; Yu, L.; McCarthy, M.P.; Marshall, J.D.; Ernst, R.K. Rationally designed TLR4 ligands for vaccine adjuvant discovery. mBio 2017, 8, e00492-17.
- Johnson, D. Synthetic TLR4-active Glycolipids as Vaccine Adjuvants and Stand-alone Immunotherapeutics. Curr. Top. Med. Chem. 2008, 8, 64–79.
- Khalaf, J.K.; Bowen, W.S.; Bazin, H.G.; Ryter, K.T.; Livesay, M.T.; Ward, J.R.; Evans, J.T.; Johnson, D.A. Characterization of TRIF selectivity in the AGP class of lipid A mimetics: Role of secondary lipid chains. Bioorg. Med. Chem. Lett 2015, 25, 547–553.
- Jiang, Z.H.; Budzynski, W.A.; Skeels, L.N.; Krantz, M.J.; Koganty, R.R. Novel lipid A mimetics derived from pentae-rythritol: Synthesis and their potent agonistic activity. Tetrahedron 2002, 58, 8833–8842.
- Akamatsu, M.; Fujimoto, Y.; Kataoka, M.; Suda, Y.; Kusumoto, S.; Fukase, K. Synthesis of lipid A monosaccharide ana-logues containing acidic amino acid: Exploring the structural basis for the endotoxic and antagonistic activities. Bioorg. Med. Chem. 2006, 14, 6759–6777.
- Ishizaka, S.T.; Hawkins, L.D. E6020: A synthetic Toll-like receptor 4 agonist as a vaccine adjuvant. Expert Rev. Vaccines 2007, 6, 773–784.
- Morefield, G.L.; Hawkins, L.D.; Ishizaka, S.T.; Kissner, T.L.; Ulrich, R.G. Synthetic Toll-like receptor 4 agonist enhances vaccine efficacy in an experimental model of toxic shock syndrome. Clin. Vaccine Immunol. 2007, 14, 1499–1504.
- Adanitsch, F.; Ittig, S.; Stöckl, J.; Oblak, A.; Haegman, M.; Jerala, R.; Beyaert, R.; Kosma, P.; Zamyatina, A. Development of aGlcN(1⇿1)aMan-based Lipid A mimetics as a novel class of potent Toll-like Receptor 4 agonists. J. Med. Chem. 2014, 57, 8056–8071.
- Adanitsch, F.; Shi, J.; Shao, F.; Beyaert, R.; Heine, H.; Zamyatina, A. Synthetic glycan-based TLR4 agonists targeting caspa-se-4/11 for the development of adjuvants and immunotherapeutics. Chem. Sci. 2018, 9, 3957–3963.
- Gordya, N.; Yakovlev, A.; Kruglikova, A.; Tulin, D.; Potolitsina, E.; Suborova, T.; Bordo, D.; Rosano, C.; Chernysh, S. Natural antimicrobial peptide complexes in the fighting of antibiotic resistant biofilms: Calliphora vicina medicinal maggots. PLoS ONE 2017, 12, e0173559.
- Chernysh, S.; Kozuharova, I. Anti-tumor activity of a peptide combining patterns of insect alloferons and mammalian immunoglobulins in naïve and tumor antigen vaccinated mice. Int. Immunopharmacol. 2013, 17, 1090–1093.
- Kim, Y.; Lee, S.K.; Bae, S.; Kim, H.; Park, Y.; Chu, N.K.; Kim, S.G.; Kim, H.-R.; Hwang, Y.-I.; Kang, J.S.; et al. The anti-inflammatory effect of alloferon on UVB-induced skin inflammation through the down-regulation of pro-inflammatory cytokines. Immunol. Lett. 2013, 149, 110–118.
- Griffin, M.E.; Hespen, C.W.; Wang, Y.; Hang, H.C. Translation of peptidoglycan metabolites into immunotherapeutics. Clin. Transl. Immunol. 2019, 8, e1095.
- Magalhaes, J.G.; Fritz, J.H.; Le Bourhis, L.; Sellge, G.; Travassos, L.H.; Selvanantham, T.; Girardin, S.E.; Gommerman, J.L.; Philpott, D.J. Nod2-Dependent Th2 Polarization of Antigen-Specific Immunity. J. Immunol. 2008, 181, 7925–7935.
- Rubino, S.J.; Magalhaes, J.G.; Philpott, D.; Bahr, G.M.; Blanot, D.; Girardin, S.E. Identification of a synthetic muramyl pep-tide derivative with enhanced Nod2 stimulatory capacity. Innate. Immun. 2013, 19, 493–503.
- Maisonneuve, C.; Bertholet, S.; Philpott, D.J.; De Gregorio, E. Unleashing the potential of NOD- and Toll-like agonists as vaccine adjuvants. Proc. Natl. Acad. Sci. USA 2014, 111, 12294–12299.
- Bumgardner, S.A.; Zhang, L.; LaVoy, A.S.; Andre, B.; Frank, C.B.; Kajikawa, A.; Klaenhammer, T.R.; Dean, G.A. Nod2 is required for antigen-specific humoral responses against antigens orally delivered using a recombinant Lactobacillus vaccine platform. PLoS ONE 2018, 13, e0196950.
- Jackson, E.M.; Herbst-Kralovetz, M.M. Intranasal Vaccination with Murabutide Enhances Humoral and Mucosal Immune Responses to a Virus-Like Particle Vaccine. PLoS ONE 2012, 7, e41529.
- Nabergoj, S.; Mlinarič-Raščan, I.; Jakopin, Ž. Harnessing the untapped potential of nucleotide-binding oligomerization domain ligands for cancer immunotherapy. Med. Res. Rev. 2019, 39, 1447–1484.
- Jakopin, Z. Murabutide Revisited: A Review of its Pleiotropic Biological Effects. Curr. Med. Chem. 2013, 20, 2068–2079.
- Telzak, E.; Wolff, S.M.; Dinarello, C.A.; Conlon, T.; El Kholy, A.; Bahr, G.M.; Choay, J.P.; Morin, A.; Chedid, L. Clinical Evaluation of the Immunoadjuvant Murabutide, a Derivative of MDP, Administered with a Tetanus Toxoid Vaccine. J. Infect. Dis. 1986, 153, 628–633.
- Przewlocki, G.; Audibert, F.; Jolivet, M.; Chedid, L.; Kent, S.B.; Neurath, A.R. Production of antibodies recognizing a hepa-titis B virus (HBV) surface antigen by administration of murabutide associated to a synthetic pre-S HBV peptide conjugated to a toxoid carrier. Biochem. Biophys. Res. Commun. 1986, 140, 557–564.
- Byars, N.E.; Nakano, G.; Welch, M.; Lehman, D.; Allison, A.C. Improvement of hepatitis B vaccine by the use of a new adju-vant. Vaccine 1991, 9, 309–318.
- Keefer, M.C.; Graham, B.S.; McElrath, M.J.; Matthews, T.J.; Stablein, D.M.; Corey, L.; Wright, P.F.; Lawrence, D.; Fast, P.E.; Weinhold, K.; et al. Safety and immunogenicity of Env 2-3, a human immunode-ficiency virus type 1 candidate vaccine, in combination with a novel adjuvant, MTP-PE/MF59. AIDS Res. Hum. Retrov. 1996, 12, 683–693.
- Tamura, M.; Yoo, Y.C.; Yoshimatsu, K.; Yoshida, R.; Oka, T.; Ohkuma, K.; Arikawa, J.; Azuma, I. Effects of muramyl dipep-tide derivatives as adjuvants on the induction of antibody response to recombinant hepatitis B surface antigen. Vaccine 1995, 13, 77–82.
- Yoo, Y.C.; Yoshimatsu, K.; Koike, Y.; Hatsuse, R.; Yamanishi, K.; Tanishita, O.; Arikawa, J.; Azuma, I. Adjuvant activity of muramyl dipeptide derivatives to enhance immunogenicity of a hantavirus-inactivated vaccine. Vaccine 1998, 16, 216–224.
- Effenberg, R.; Turánek Knötigová, P.; Zyka, D.; Célechovská, H.; Mašek, J.; Bartheldyová, E.; Hubatka, F.; Koudelka, S.; Lukáč, R.; Kovalová, A.; et al. Nonpyrogenic molecular adjuvants based on norAbu-muramyldipeptide and norAbu-glucosaminyl muramyldipeptide: Synthesis, molecular mechanisms of action, and biological activities in vitro and in vivo. J. Med. Chem. 2017, 60, 7745–7763.
- Yang, H.Z.; Xu, S.; Liao, X.Y.; Zhang, S.D.; Liang, Z.L.; Liu, B.H.; Bai, J.Y.; Jiang, C.; Ding, J.; Cheng, G.F.; et al. A Novel Immunostimulator, N2-[α-O-Benzyl-N-(acetylmuramyl)-l-alanyl-d- isoglutaminyl]-N6-trans-(m-nitrocinnamoyl)-l-lysine, and Its Adjuvancy on the Hepatitis B Surface Antigen. J. Med. Chem. 2005, 48, 5112–5122.
- Gobec, M.; Tomašič, T.; Štimac, A.; Frkanec, R.; Trontelj, J.; Anderluh, M.; Mlinarič-Raščan, I.; Jakopin, Ž. Discovery of na-nomolar desmuramylpeptide agonists of the innate immune receptor nucleotide-binding oligomerization domain-containing protein 2 (NOD2) possessing immunostimulatory properties. J. Med. Chem. 2018, 61, 2707–2724.
- Fernández-Tejada, A.; Tan, D.S.; Gin, D.Y. Development of Improved Vaccine Adjuvants Based on the Saponin Natural Product QS-21 through Chemical Synthesis. Acc. Chem. Res. 2016, 49, 1741–1756.
- Pifferi, C.; Fuentes, R.; Fernández-Tejada, A. Natural and synthetic carbohydrate-based vaccine adjuvants and their mechanisms of action. Nat. Rev. Chem. 2021, 5, 197–216.
- Fernández-Tejada, A. Design, synthesis and evaluation of optimized saponin variants derived from the vaccine adjuvant QS-21. Pure Appl. Chem. 2017, 89, 1359–1378.
- Fuentes, R.; Ruiz-de-Angulo, A.; Sacristán, N.; Navo, C.D.; Jiménez-Osés, G.; Anguita, J.; Fernández-Tejada, A. Replacing the Rhamnose-Xylose Moiety of QS-21 with Simpler Terminal Disaccharide Units Attenuates Adjuvant Activity in Truncated Saponin Variants. Chem. Eur. J. 2021, 27, 4731–4737.
- Fernández-Tejada, A.; Chea, E.K.; George, C.; Pillarsetty, N.V.K.; Gardner, J.R.; Livingston, P.O.; Ragupathi, G.; Lewis, J.S.; Tan, D.S.; Gin, D.Y. Development of a minimal saponin vaccine adjuvant based on QS-21. Nat. Chem. 2014, 6, 635–643.
- Ghirardello, M.; Ruiz-de-Angulo, A.; Sacristan, N.; Barriales, D.; Jiménez-Barbero, J.; Poveda, A.; Corzana, F.; Anguita, J.; Fernández-Tejada, A. Exploting structure–activity relationships of QS-21 in the design and synthesis of streamlined saponin vaccine adjuvants. Chem. Commun. 2020, 56, 719–722.
- Le, T.T.; Cramer, J.P.; Chen, R.; Mayhew, S. Evolution of the COVID-19 vaccine development landscape. Nat. Rev. Drug Discov. 2020, 19, 667–668.
- Jeyanathan, M.; Afkhami, S.; Smaill, F.; Miller, M.S.; Lichty, B.D.; Xing, Z. Immunological considerations for COVID-19 vaccine strategies. Nat. Rev. Immunol. 2020, 20, 1–18.
- Huang, X.; Wang, X.; Zhang, J.; Xia, N.; Zhao, Q. Escherichia coli-derived virus-like particles in vaccine development. Npj Vaccines 2017, 2, 1–9.
- Donaldson, B.; Lateef, Z.; Walker, G.F.; Young, S.L.; Ward, V.K. Virus-like particle vaccines: Immunology and formula-tion for clinical translation. Expert. Rev. Vaccines 2018, 17, 833–849.
- Ward, B.J.; Gobeil, P.; Séguin, A.; Atkins, J.; Boulay, I.; Charbonneau, P.Y.; Couture, M.; D’Aoust, M.-A.; Dhaliwall, J.; Finkle, C.; et al. Phase 1 trial of a Candidate Recombinant Virus-Like Particle Vaccine for Covid-19 Disease Produced in Plants. medRxiv 2020.
- Gursel, M.; Gursel, I. Development of CpG ODN Based Vaccine Adjuvant Formulations. Adv. Struct. Saf. Stud. 2016, 1404, 289–298.
- Bode, C.; Zhao, G.; Steinhagen, F.; Kinjo, T.; Klinman, D.M. CpG DNA as a vaccine adjuvant. Expert Rev. Vaccines 2011, 10, 499–511.
- Klimek, L.; Willers, J.; Hammann-Haenni, A.; Pfaar, O.; Stocker, H.; Mueller, P.; Renner, W.A.; Bachmann, M.F. Assessment of clinical efficacy of CYT003-QbG10 in patients with allergic rhinoconjunctivitis: A phase IIb study. Clin. Exp. Allergy 2011, 41, 1305–1312.


