 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Bernard Lopez | + 1532 word(s) | 1532 | 2021-04-27 07:56:08 | | | |
| 2 | Bruce Ren | -21 word(s) | 1511 | 2021-04-28 04:35:51 | | |
Video Upload Options
Homologous recombination (HR) is a fundamental evolutionarily conserved process that plays prime role(s) in genome stability maintenance through DNA repair and through the protection and resumption of arrested replication forks. HR promotes the exchange between homologous DNA sequences resulting in a novel combination of the genetic material. Therefore, HR is essential in genome stability maintenance but also plays an important role in genome diversity; such as in the case of meiosis. Many HR genes are deregulated in cancer cells. Notably, the breast cancer genes BRCA1 and BRCA2, two important HR players, are the most frequently mutated genes in familial breast and ovarian cancer.
1. Introduction
Homologous recombination (HR), promotes the exchange between homologous DNA sequences resulting in a novel combination of the genetic material, is a molecular process highly conserved through evolution that plays prominent roles in genome plasticity. Indeed, the DNA repair function(s) and the outcomes of HR are essential not only to maintain genome stability but also to promote genome variability [1][2][3][4][5]. HR allows the repair of different types of DNA lesions, mainly DNA double-strand breaks (DSBs) and interstrand crosslinks (ICLs) [6][7]. Moreover, an important role of HR in genome stability maintenance is the protection and resumption of arrested replication forks. However, prolonged replication fork arrest leads to DSBs that can thus be processed by HR [8][9].
DSBs are generally considered the most toxic lesion. DSBs can be generated by endogenous stresses resulting from cellular metabolism, such as replication stress and reactive oxygen species (ROS), as well as from exogenous factors, such as ionizing radiation and chemotherapy agents (e.g., topoisomerase inhibitors). DSBs can also be programmed to trigger beneficial genomic rearrangements during meiotic differentiation [10] or the establishment of the immune system [11]. HR is also a driving force for the evolution of multigene families [12]. Therefore, thanks to this versatility, HR is involved in many fundamental biological processes. Finally, the technical application of HR constitutes the basis of targeted gene replacement for gene therapy as well as for the precise design of engineered organisms [13][14].
Genetic instability is a hallmark of aging and cancer [15][16][17]. Remarkably, markers of the DNA damage response have been found to be activated at pre-/early steps of tumorigenesis [18][19]. Since replication stress is a prominent endogenous source of DNA damage and genome instability, these data indicate a causal role of DNA replication stress in the early steps of tumor initiation [18][19][20]. Note that genetic instability can also fuel tumor progression. Because of its essential roles in genome stability maintenance, particularly in response to replication stress, HR is generally considered a tumor suppressor mechanism. Indeed, germline and somatic inactivating mutations in major HR actors have been observed in different types of tumors [21]. For example, germline heterozygous mutations in genes directly implicated in HR—BRCA1, BRCA2, PALB2, RAD51C and RAD51D—increase the risk of ovarian and breast cancer [22][23][24][25]. In addition, Fanconi anemia patients with biallelic mutations in BRCA1, BRCA2 and PALB2 show an increased risk of hematological malignancies and solid tumors [26][27][28][29][30][31][32][33].
2. Molecular Mechanisms of HR
HR-mediated DSB repair represents a paradigm for the molecular steps of HR. DSBs are first recognized and signaled by the MRN complex (MRE11/RAD50/NBS1) in cooperation with the ATM (ataxia-telangiectasia mutated) kinase and the chromatin remodeling machinery. Subsequently, DSBs can be either repaired by canonical nonhomologous end-joining (cNHEJ) or resected (Figure 1A) to then be repaired by HR or other resection-dependent DSB repair processes, such as single strand annealing (SSA) and alternative end-joining [34][35][36]. This choice between NHEJ and resection is controlled by the antagonistic activities of BRCA1, which favors resection, in competition with the protein complex 53BP1-Shieldin (53BP1-REV7-SHLD1-SHLD2-SHLD3), which prevents DNA end resection [37][38][39] (Figure 1A). The steps of HR that succeed DSB recognition can be summarized as follows (Figure 1B): (1) DSB resection to generate a 3′ ssDNA stretch (see Figure 1A); (2) RAD51 loading on the ssDNA, forming the ssDNA-RAD51 filament that promotes a search for homology and invasion of a homologous duplex DNA; (3) DNA synthesis primed by the invading 3′ end; and (4) the formation and resolution of HR intermediates that give rise to gene conversions (nonreciprocal exchange of genetic information) either associated with crossover or not (reciprocal exchange of adjacent sequences) (Figure 1B) (reviewed in [1][3][5]).
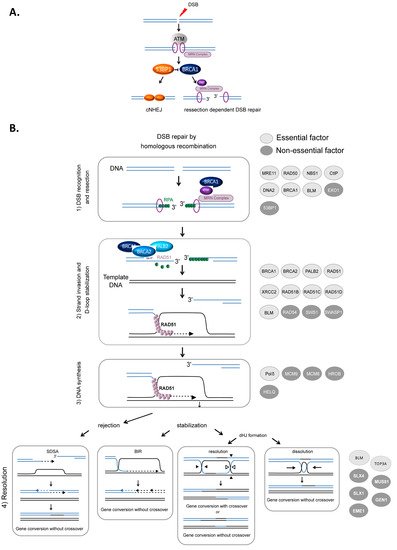
Figure 1. HR-mediated DSB repair. (A). Signaling and competition between cNHEJ and resection [34]. The involvement of 53BP1, BRCA1 and the MRN complex. (B). Model of DSB repair by HR. (1) The MRN complex initiates a 5′ to 3′ resection under the control of BRCA1/CtIP, generating a 3′ single-stranded DNA (ssDNA) (A). (2) RAD51 is then loaded to the 3′ single-stranded DNA by the combined action of BRCA2/PALB2 and BRCA1, resulting in the formation of an ordered RAD51-ssDNA nucleofilament. The invasion of homologous duplex DNA by the RAD51-ssDNA filament induces the formation of a displacement loop (D-loop). (3) The invading strand primes DNA synthesis. (4) The D-loop can be either dismantled, leading to DSB repair by synthesis-dependent strand annealing (SDSA) or stabilized, leading to either DSB repair by BIR or the formation of a double Holliday junction (dHJ). The dHJ can be resolved or dissolved, leading to a repair product either associated with a crossover event or not. The proteins involved in each corresponding step are listed on the right side of the figure. Light gray circles indicate essential factors; essential factors are defined as genes whose knockout leads to embryonic lethality in mice. Dark gray circles indicate nonessential factors; these are genes whose KO does not lead to embryonic lethality in mice.
2.1. Resection
Resection, which is required to initiate HR, is performed in two steps (Figure 1A). BRCA1 is recruited to DSB sites via its interaction with NBS1 and associates with BARD1, forming an active E3 ubiquitin ligase. This complex ubiquitinates the endonuclease CtIP, which cooperates with MRE11 to initiate DSB resection [40][41]. Then, exonuclease 1 (EXO1) and/or the BLM/DNA2 (helicase/nuclease) complex extend the 3′ overhang [42][43][44][45] (Figure 1B). Finally, the 3′ ssDNA stretch created by resection is coated with replication protein A (RPA), protecting it (Figure 1B).
2.2. Loading RAD51 on ssDNA, Search for Homology and Strand Invasion
The loading of RAD51 onto ssDNA is performed by the BRCA2-PALB2 complex [46][47]. This protein complex interacts with BRCA1 and catalyzes the replacement of RPA by RAD51 on the stretch of 3′ ssDNA, creating the RAD51-ssDNA presynaptic complex [48][49][50]. Note that BRCA1 plays roles during different steps of HR: the initiation of resection and the loading of RAD51 (Figure 1B) [3][51][52].
The ssDNA-RAD51 filament scans the genome to search for homology. Once a homologous sequence is found, the filament invades the duplex homologous DNA and initiates strand exchange, creating a displacement loop (D-loop).
2.3. DNA Synthesis
The 3′ invading strand primes DNA synthesis through the recruitment of DNA and the copy of the invaded DNA molecule. Numerous studies have demonstrated the involvement of many polymerases in this process, although Polδ has been proposed to play a primary role [53]. The protein complexes HROB-MCM8–MCM9 and HELQ are proposed to have redundant helicase functions to promote DNA synthesis during HR [54].
2.4. Formation and Resolution of HR Intermediates
Strand invasion and DNA synthesis lead to the formation of different intermediates whose processing leads to gene conversion either associated with crossover products or not (Figure 1). The invading strand can be disassembled, channeling DSB repair toward synthesis-dependent strand annealing (SDSA) (Figure 1B). If stabilized, the D-loop can lead to DSB repair by break-induced repair (BIR) or to the formation of double Holliday junctions that can be either dissolved by the BLM-TOP3A-RMI1/2 complex or resolved by the structure-specific resolvases MUS81-EME1, GEN1 or SLX1 (Figure 1B) (reviewed in [2][55]).
Of note, the DNA helicase BLM, which is mutated in Bloom syndrome, plays several roles, sometimes contradictory, and at different HR steps. Indeed, BLM is involved in different steps of HR, including end resection at HR initiation [42][56], D-loop rejection and dHJ resolution at HR termination [56][57]. At resection initiation, depending on the cell cycle phase that modifies its interacting partners, BLM either favors the loading of 53BP1 on the DSB in G1 phase, preventing the initiation of unscheduled resection, or, in contrast, favors resection in S phase when interacting with TOP3 [58].
2.5. Accessory Proteins
RAD54, a member of the SWI2/SNF2 protein family (ATP-dependent chromatin remodelers), interacts with RAD51, and in vitro studies have proposed that it functions as a RAD51 cofactor [59][60][61]. RAD54 catalyzes the extension of joint molecules [62] and stabilizes the D-loop [63].
A family of six proteins (RAD51B, RAD51C, RAD51D, XRCC2, XRCC3, and RAD51AP1), known as the RAD51 paralogs (i.e., proteins that share sequence homology with RAD51 in a given species), has been identified in mammals. Two distinct complexes have been identified: RAD51B–RAD51C–RAD51D–XRCC2 (BCDX2) and RAD51C–XRCC3 (CX3) [64]. RAD51 paralogs favor the recruitment of RAD51 to DNA damage sites [65] and promote the formation and stabilization of the RAD51 nucleoprotein filament. However, the exact role of each paralog remains to be fully determined. In addition, RAD51 paralogs influence gene conversion tract length [66][67]. The SWSAP1 protein, a noncanonical paralog of RAD51, forms the so-called SHU complex when associated with SWS1 (SWSAP1-SWS1). SHU interacts with RAD51 and regulates its function [68].
References
- Haber, J.E. Genome Stability. DNA Repair and Recombination, 1st ed.; Garland Science: New York, NY, USA, 2014; ISBN 1317682319.
- Kowalczykowski, S.C. An overview of the molecular mechanismsof recombinational DNA repair. Cold Spring Harb. Perspect. Biol. 2015, 7, a016410.
- Jasin, M.; Rothstein, R. Repair of strand breaks by homologous recombination. Cold Spring Harb. Perspect. Biol. 2013, 5, a012740.
- Piazza, A.; Heyer, W.D. Homologous Recombination and the Formation of Complex Genomic Rearrangements. Trends Cell Biol. 2019, 29, 135–149.
- Wright, W.D.; Shah, S.S.; Heyer, W.D. Homologous recombination and the repair of DNA double-strand breaks. J. Biol. Chem. 2018, 293, 10524–10535.
- Deans, A.J.; West, S.C. DNA interstrand crosslink repair and cancer. Nat. Rev. Cancer 2011, 11, 467–480.
- Chen, C.-C.; Feng, W.; Lim, P.X.; Kass, E.M.; Jasin, M. Homology-Directed Repair and the Role of BRCA1, BRCA2, and Related Proteins in Genome Integrity and Cancer. Annu. Rev. Cancer Biol. 2018, 2, 313–336.
- Petermann, E.; Orta, M.L.; Issaeva, N.; Schultz, N.; Helleday, T. Hydroxyurea-stalled replication forks become progressively inactivated and require two different RAD51-mediated pathways for restart and repair. Mol. Cell 2010, 37, 492–502.
- Saintigny, Y.; Delacote, F.; Vares, G.; Petitot, F.; Lambert, S.; Averbeck, D.; Lopez, B.S. Characterization of homologous recombination induced by replication inhibition in mammalian cells. EMBO J. 2001, 20, 3861–3870.
- Hunter, N. Meiotic recombination: The essence of heredity. Cold Spring Harb. Perspect. Biol. 2015, 7, a016618.
- Jung, D.; Alt, F.W. Unraveling V(D)J recombination; insights into gene regulation. Cell 2004, 116, 299–311.
- Guirouilh-Barbat, J.; Lambert, S.; Bertrand, P.; Lopez, B.S. Is homologous recombination really an error-free process? Front. Genet. 2014, 5, 175.
- Rocha-Martins, M.; Cavalheiro, G.R.; Matos-Rodrigues, G.E.; Martins, R.A.P. From gene targeting to genome editing: Transgenic animals applications and beyond. An. Acad. Bras. Cienc. 2015, 87, 1323–1348.
- Gaj, T.; Gersbach, C.A.; Barbas, C.F. ZFN, TALEN, and CRISPR/Cas-based methods for genome engineering. Trends Biotechnol. 2013, 31, 397–405.
- Negrini, S.; Gorgoulis, V.G.; Halazonetis, T.D. Genomic instability—An evolving hallmark of cancer. Nat. Rev. Mol. Cell Biol. 2010, 11, 220–228.
- López-Otín, C.; Blasco, M.A.; Partridge, L.; Serrano, M.; Kroemer, G. The hallmarks of aging. Cell 2013, 153, 1194–1217.
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674.
- Bartkova, J.; Horejsi, Z.; Koed, K.; Kramer, A.; Tort, F.; Zieger, K.; Guldberg, P.; Sehested, M.; Nesland, J.M.; Lukas, C.; et al. DNA damage response as a candidate anti-cancer barrier in early human tumorigenesis. Nature 2005, 434, 864–870.
- Gorgoulis, V.G.; Vassiliou, L.V.; Karakaidos, P.; Zacharatos, P.; Kotsinas, A.; Liloglou, T.; Venere, M.; Ditullio, R.A.J.; Kastrinakis, N.G.; Levy, B.; et al. Activation of the DNA damage checkpoint and genomic instability in human precancerous lesions. Nature 2005, 434, 907–913.
- Tubbs, A.; Nussenzweig, A. Endogenous DNA Damage as a Source of Genomic Instability in Cancer. Cell 2017, 168, 644–656.
- Lord, C.J.; Ashworth, A. BRCAness revisited. Nat. Rev. Cancer 2016, 16, 110–120.
- Kuchenbaecker, K.B.; Hopper, J.L.; Barnes, D.R.; Phillips, K.; Mooij, T.M.; Jervis, S.; Van Leeuwen, F.E.; Milne, R.L.; Andrieu, N.; Goldgar, D.E.; et al. Risks of Breast, Ovarian, and Contralateral Breast Cancer for. JAMA 2017, 317, 2402–2416.
- Antoniou, A.C.; Casadei, S.; Heikkinen, T.; Barrowdale, D.; Pylkäs, K.; Roberts, J.; Lee, A.; Subramanian, D.; De Leeneer, K.; Fostira, F.; et al. Breast-cancer risk in families with mutations in PALB2. N. Engl. J. Med. 2014, 371, 497–506.
- Coulet, F.; Fajac, A.; Colas, C.; Eyries, M.; Dion-Minière, A.; Rouzier, R.; Uzan, S.; Lefranc, J.P.; Carbonnel, M.; Cornelis, F.; et al. Germline RAD51C mutations in ovarian cancer susceptibility. Clin. Genet. 2013, 83, 332–336.
- Ducy, M.; Sesma-Sanz, L.; Guitton-Sert, L.; Lashgari, A.; Gao, Y.; Brahiti, N.; Rodrigue, A.; Margaillan, G.; Caron, M.C.; Côté, J.; et al. The Tumor Suppressor PALB2: Inside Out. Trends Biochem. Sci. 2019, 44, 226–240.
- Kee, Y.; D’Andrea, A.D. Molecular pathogenesis and clinical management of Fanconi anemia. J. Clin. Investig. 2012, 122, 3799–3806.
- Domchek, S.M.; Tang, J.; Stopfer, J.; Lilli, D.R.; Hamel, N.; Tischkowitz, M.; Monteiro, A.N.A.; Messick, T.E.; Powers, J.; Yonker, A.; et al. Biallelic deleterious BRCA1 mutations in a woman with early-onset ovarian cancer. Cancer Discov. 2013, 3, 399–405.
- Sawyer, S.L.; Tian, L.; Kähkönen, M.; Schwartzentruber, J.; Kircher, M.; Majewski, J.; Dyment, D.A.; Innes, A.M.; Boycott, K.M.; Moreau, L.A.; et al. Biallelic mutations in BRCA1 cause a new Fanconi anemia subtype. Cancer Discov. 2015, 5, 135–142.
- Freire, B.L.; Homma, T.K.; Funari, M.F.A.; Lerario, A.M.; Leal, A.M.; Velloso, E.D.R.P.; Malaquias, A.C.; Jorge, A.A.L. Homozygous loss of function BRCA1 variant causing a Fanconi-anemia-like phenotype, a clinical report and review of previous patients. Eur. J. Med. Genet. 2018, 61, 130–133.
- Niraj, J.; Färkkilä, A.; D’Andrea, A.D. The Fanconi Anemia Pathway in Cancer. Annu. Rev. Cancer Biol. 2019, 3, 457–478.
- Howlett, N.G.; Taniguchi, T.; Olson, S.; Cox, B.; Waisfisz, Q.; De Die-Smulders, C.; Persky, N.; Grompe, M.; Joenje, H.; Pals, G.; et al. Biallelic inactivation of BRCA2 in Fanconi anemia. Science 2002, 297, 606–609.
- Seo, A.; Orna, S.S.; Unal, S.; Casadei, S.; Walsh, T.; Gumruk, F.; Shalev, S.; Shimamura, A.; Akarsu, N.A.; Tamary, H.; et al. Mechanism for survival of homozygous nonsense mutations in the tumor suppressor gene BRCA1. Proc. Natl. Acad. Sci. USA 2018, 115, 5241–5246.
- Auerbach, A.D. Fanconi anemia and its diagnosis. Mutat. Res. 2009, 668, 4–10.
- Rass, E.; Grabarz, A.; Plo, I.; Gautier, J.; Bertrand, P.; Lopez, B.S.B.S. Role of Mre11 in chromosomal nonhomologous end joining in mammalian cells. Nat. Struct. Mol. Biol. 2009, 16, 819–824.
- Betermier, M.; Bertrand, P.; Lopez, B.S.B.S.; Bétermier, M.; Bertrand, P.; Lopez, B.S.B.S. Is Non-Homologous End-Joining Really an Inherently Error-Prone Process? PLoS Genet. 2014, 10, e1004086.
- So, A.; Le Guen, T.; Lopez, B.S.B.S.; Guirouilh-Barbat, J. Genomic rearrangements induced by unscheduled DNA double strand breaks in somatic mammalian cells. FEBS J. 2017, 284, 2324–2344.
- Mirman, Z.; de Lange, T. 53BP1: A DSB escort. Genes Dev. 2020, 34, 7–23.
- Setiaputra, D.; Durocher, D. Shieldin—The protector of DNA ends. EMBO Rep. 2019, 20, e47560.
- Tarsounas, M.; Sung, P. The antitumorigenic roles of BRCA1–BARD1 in DNA repair and replication. Nat. Rev. Mol. Cell Biol. 2020, 21, 284–299.
- Yu, X.; Fu, S.; Lai, M.; Baer, R.; Chen, J. BRCA1 ubiquitinates its phosphorylation-dependent binding partner CtIP. Genes Dev. 2006, 20, 1721–1726.
- Sartori, A.A.; Lukas, C.; Coates, J.; Mistrik, M.; Fu, S.; Bartek, J.; Baer, R.; Lukas, J.; Jackson, S.P. Human CtIP promotes DNA end resection. Nature 2007, 450, 509–514.
- Nimonkar, A.V.; Genschel, J.; Kinoshita, E.; Polaczek, P.; Campbell, J.L.; Wyman, C.; Modrich, P.; Kowalczykowski, S.C. BLM-DNA2-RPA-MRN and EXO1-BLM-RPA-MRN constitute two DNA end resection machineries for human DNA break repair. Genes Dev. 2011, 25, 350–362.
- Mimitou, E.P.; Symington, L.S. Sae2, Exo1 and Sgs1 collaborate in DNA double-strand break processing. Nature 2008, 455, 770–774.
- Zhu, Z.; Chung, W.H.; Shim, E.Y.; Lee, S.E.; Ira, G. Sgs1 helicase and two nucleases Dna2 and Exo1 resect DNA double-strand break ends. Cell 2008, 134, 981–994.
- Cejka, P.; Cannavo, E.; Polaczek, P.; Masuda-Sasa, T.; Pokharel, S.; Campbell, J.L.; Kowalczykowski, S.C. DNA end resection by Dna2-Sgs1-RPA and its stimulation by Top3-Rmi1 and Mre11-Rad50-Xrs2. Nature 2010, 467, 112–116.
- Jensen, R.B.; Carreira, A.; Kowalczykowski, S.C. Purified human BRCA2 stimulates RAD51-mediated recombination. Nature 2010, 467, 678–683.
- Thorslund, T.; McIlwraith, M.J.; Compton, S.A.; Lekomtsev, S.; Petronczki, M.; Griffith, J.D.; West, S.C. The breast cancer tumor suppressor BRCA2 promotes the specific targeting of RAD51 to single-stranded DNA. Nat. Struct. Mol. Biol. 2010, 17, 1263–1265.
- Xia, B.; Sheng, Q.; Nakanishi, K.; Ohashi, A.; Wu, J.; Christ, N.; Liu, X.; Jasin, M.; Couch, F.J.; Livingston, D.M. Control of BRCA2 Cellular and Clinical Functions by a Nuclear Partner, PALB2. Mol. Cell 2006, 22, 719–729.
- Sy, S.M.H.; Huen, M.S.Y.; Chen, J. PALB2 is an integral component of the BRCA complex required for homologous recombination repair. Proc. Natl. Acad. Sci. USA 2009, 106, 7155–7160.
- Zhang, F.; Ma, J.; Wu, J.; Ye, L.; Cai, H.; Xia, B.; Yu, X. PALB2 Links BRCA1 and BRCA2 in the DNA-Damage Response. Curr. Biol. 2009, 19, 524–529.
- Zhao, W.; Wiese, C.; Kwon, Y.; Hromas, R.; Sung, P. The BRCA tumor suppressor network in chromosome damage repair by homologous recombination. Annu. Rev. Biochem. 2019, 88, 221–245.
- Moynahan, M.E.; Chiu, J.W.; Koller, B.H.; Jasin, M. Brca1 controls homology-directed DNA repair. Mol. Cell 1999, 4, 511–518.
- McVey, M.; Khodaverdian, V.Y.; Meyer, D.; Cerqueira, P.G.; Heyer, W.-D. Eukaryotic DNA Polymerases in Homologous Recombination. Annu. Rev. Genet. 2016, 50, 393–421.
- Hustedt, N.; Saito, Y.; Zimmermann, M.; Álvarez-Quilón, A.; Setiaputra, D.; Adam, S.; McEwan, A.; Yuan, J.Y.; Olivieri, M.; Zhao, Y.; et al. Control of homologous recombination by the HROB-MCM8-MCM9 pathway. Genes Dev. 2019, 33, 1397–1415.
- Bizard, A.H.; Hickson, I.D. The dissolution of double Holliday junctions. Cold Spring Harb. Perspect. Biol. 2014, 6, a016477.
- Chu, W.K.; Hanada, K.; Kanaar, R.; Hickson, I.D. BLM has early and late functions in homologous recombination repair in mouse embryonic stem cells. Oncogene 2010, 29, 4705–4714.
- Larsen, N.B.; Hickson, I.D. RecQ helicases: Conserved guardians of genomic integrity. Adv. Exp. Med. Biol. 2013, 767, 161–184.
- Grabarz, A.; Guirouilh-Barbat, J.J.; Barascu, A.A.; Pennarun, G.G.; Genet, D.; Rass, E.; Germann, S.M.S.M.S.M.; Bertrand, P.; Hickson, I.D.I.D.I.D.; Lopez, B.S.B.S. A role for BLM in double-strand break repair pathway choice: Prevention of CtIP/Mre11-mediated alternative nonhomologous end-joining. Cell Rep. 2013, 5, 21–28.
- Mazin, A.V.; Bornarth, C.J.; Solinger, J.A.; Heyer, W.D.; Kowalczykowski, S.C. Rad54 protein is targeted to pairing loci by the Rad51 nucleoprotein filament. Mol. Cell 2000, 6, 583–592.
- Crickard, J.B.; Moevus, C.J.; Kwon, Y.; Sung, P.; Greene, E.C. Rad54 Drives ATP Hydrolysis-Dependent DNA Sequence Alignment during Homologous Recombination. Cell 2020, 181, 1380–1394.e18.
- Sigurdsson, S.; Van Komen, S.; Petukhova, G.; Sung, P. Homologous DNA pairing by human recombination factors Rad51 and Rad54. J. Biol. Chem. 2002, 277, 42790–42794.
- Bugreev, D.V.; Hanaoka, F.; Mazin, A.V. Rad54 dissociates homologous recombination intermediates by branch migration. Nat. Struct. Mol. Biol. 2007, 14, 746–753.
- Wright, W.D.; Heyer, W.D. Rad54 Functions as a Heteroduplex DNA Pump Modulated by Its DNA Substrates and Rad51 during D Loop Formation. Mol. Cell 2014, 53, 420–432.
- Masson, J.Y.; Tarsounas, M.C.; Stasiak, A.Z.; Stasiak, A.; Shah, R.; McIlwraith, M.J.; Benson, F.E.; West, S.C. Identification and purification of two distinct complexes containing the five RAD51 paralogs. Genes Dev. 2001, 15, 3296–3307.
- Garcin, E.B.; Gon, S.; Sullivan, M.R.; Brunette, G.J.; de Cian, A.; Concordet, J.P.; Giovannangeli, C.; Dirks, W.G.; Eberth, S.; Bernstein, K.A.; et al. Differential requirements for the RAD51 paralogs in genome repair and maintenance in human cells. PLoS Genet. 2019, 15, e1008355.
- Nagaraju, G.; Odate, S.; Xie, A.; Scully, R. Differential Regulation of Short- and Long-Tract Gene Conversion between Sister Chromatids by Rad51C. Mol. Cell. Biol. 2006, 26, 8075–8086.
- Nagaraju, G.; Hartlerode, A.; Kwok, A.; Chandramouly, G.; Scully, R. XRCC2 and XRCC3 Regulate the Balance between Short- and Long-Tract Gene Conversions between Sister Chromatids. Mol. Cell. Biol. 2009, 29, 4283–4294.
- Sullivan, M.R.; Bernstein, K.A. RAD-ical new insights into RAD51 regulation. Genes 2018, 9, 629.

