 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Sandrine Herbelet | + 4130 word(s) | 4130 | 2021-04-20 04:21:41 | | | |
| 2 | Conner Chen | Meta information modification | 4130 | 2021-04-26 11:28:08 | | |
Video Upload Options
In Duchenne muscular dystrophy (DMD), the absence of dystrophin from the dystrophin-associated protein complex (DAPC) causes muscle membrane instability, which leads to myofiber necrosis, hampered regeneration, and chronic inflammation. The resulting disabled DAPC-associated cellular pathways have been described both at the molecular and the therapeutical level, with the Toll-like receptor nuclear factor kappa-light-chain-enhancer of activated B cells pathway (NF-ƘB), Janus kinase/signal transducer and activator of transcription proteins, and the transforming growth factor-β pathways receiving the most attention. The protein kinase A/ mitogen-activated protein kinase/nuclear factor of activated T-cells 5/organic osmolytes (PKA-p38MAPK-NFAT5-organic osmolytes) pathway. This pathway plays an important role in osmotic homeostasis essential to normal cell physiology via its regulation of the influx/efflux of organic osmolytes. Besides, NFAT5 plays an essential role in cell survival under hyperosmolar conditions, in skeletal muscle regeneration, and in tissue inflammation, closely interacting with the master regulator of inflammation NF-ƘB.
1.Duchenne Muscular Dystrophy and PKA-p38MAPK-NFAT5-Pathway
Duchenne muscular dystrophy (DMD) is a severe X-linked disorder usually recognized in early childhood, characterized by progressive skeletal muscle weakness causing loss of ambulation by early adolescence. In addition to the skeletal muscle, the disorder also involves cardiac and respiratory muscles. DMD is caused by disruptive mutations in the DMD gene, resulting in the absence of its protein product dystrophin from muscle fiber membranes. Contraction damages the dystrophin-deficient muscle fibers, generating cycles of muscle fiber necrosis and regeneration that fail to restore the tissue, leading to fibrosis and fatty replacement. Absence of dystrophin distorts anchoring of the extracellular matrix to the myofiber cytoskeleton [1][2][3]. Dystrophin is part of a larger group of transmembrane proteins, called the dystrophin associated protein complex (DAPC), which has the ability to receive and transduce signals from in- and outside the myofiber and absorb shocks during muscle contraction [4][5]. This shock-absorbing effect of the DAPC is compromised in DMD patients and causes cell membrane instability, which start as soon as the fetus can move in utero. The DAPC is composed of dystrobrevins, dystroglycans, sarcoglycans, sarcospan, and syntrophin. The most widely used murine model for DMD are mdx mice that carry a nonsense mutation in exon 23 of the dystrophin gene. However, the disease phenotype of this murine model is milder than the human condition. While humans display progressive muscle weakness, mice develop active muscle tissue degeneration and regeneration at a young age, which later on continues at a slower pace. The DAPC is affected in DMD and mdx displaying both common and differential deficiencies (Table 1). In mdx mice, α- and β-dystroglycans are unstable, whereas expression of α-, β-, γ- and δ-sarcoglycans, sarcospan and α1-syntrophin is weak [6][7][8][9][10][11] and expression of β1-syntrophin and α-dystrobrevin is absent [12][13][14]. In DMD patients, the following DAPC proteins are less expressed to sometimes absent: α- dystroglycan, α-sarcoglycan, sarcospan, α1-syntrophin, α-dystrobrevin, and neuronal nitric oxide synthase (nNOS) [10][15][16][17][18][19][20][21][22]. The role of nNOS instability in mdx pathology is controversial. Indeed, normal nNOS activity reduced dystrophic symptoms in one mdx study, whereas mdx mice crossed with NOS-null mice showed no difference in muscle pathology when compared to mdx mice [23][24][25]. β-dystroglycan is still present in DMD skeletal muscle tissue [6] and expression studies of β1-syntrophin in DMD patients are not yet available. Both β-dystroglycan and syntrophin function as signaling proteins. More specifically, β-dystroglycan signals to Ras-related C3 botulinum toxin substrate 1 (Rac1) small guanosinetrifosfaat (GTP)ase and to mitogen-activated protein kinase (MAPK) through growth factor receptor-bound protein 2 (Grb2). Syntrophin organizes a signalplex linked to dystrophin and regulates signaling proteins such as voltage-gated sodium channels along with plasma membrane calcium pumps and nNOS. DAPC also interacts with calmodulin which is stimulated by calcium and in turn signals to calcineurin [5][26][27].
Table 1. Differential expression of the main dystrophin-associated protein complex (DAPC) components in Duchenne muscular dystrophy (DMD) and in the mdx mouse.
| Organic Osmolyte | Physiological Role | Expression in DMD Patients/mdx Mouse | Clinical Trials |
|---|---|---|---|
| Taurine | Osmoregulation, antioxidant, modulation of Ca2+ signaling, conjugation of bile acids |
DMD: ↑ taurine excretion ↑ taurine trans-porter (TauT) mdx: ↓ muscle taurine ↓ TauT |
Peripartum cardiomyopathy, thalassemia major, diabetes, etc. |
| Betaine | Osmoregulation, methyldonor |
DMD: expression in small regenerative and atrophic fibers | Homocystinuria, non-alcoholic fatty liver disease, etc. |
| Myo-inositol (MI) | Osmoregulation, structural base for 2nd messengers (signal transduction, component of plasma membrane) | DMD: ↑ sodium/myo-inositol transporter (SMIT) | Polycystic ovary syndrome, bipolar disorders, etc. |
| Sorbitol | Osmoregulation | DMD: ↑ Aldose reductase mdx: ↑ Aldose reductase (cardiac muscle) |
Hampered signal transduction and cellular pathways have been described in DMD with Toll-like receptor/tumor necrosis factor α (TNF-α)/interleukin 1β (IL-1β)/interleukin 6 (IL-6)-nuclear factor kappa-light-chain-enhancer of the activated B cell pathway (NF-ƘB), Janus kinase/signal transducer and activator of transcription proteins, and the transforming growth factor-β (TGF-β) pathways having been extensively studied and considered for therapeutic targeting [28][29]. Due to the important inflammatory aspect of the disease, glucocorticoids (GCs) are the drugs of choice in DMD with their major mode of action residing in the binding to NF-ƘB and MAPKs along with nuclear translocation of nuclear factor of activated T-cells (NFAT) [30][31]. The NFAT group consists of five transcription factors belonging to the larger ReI family which also encompasses NF-ƘB. NFATc1-4 are regulated by calcineurin, whereas NFAT5 is a non-calcineurin mediated transcription factor harboring similarities with both NFATc and NF-ƘB [32]. NFAT5 is a multifaceted protein, which tightly controls cell volume in order to remain inside the homeostatic range. It controls cell growth in embryogenic tissue, mediates inflammation and protects cells from oxidative stress and metabolic aberrations due for instance to excessive caloric intake. It therefore deserves the name of immunometabolic stress protein [33].
In DMD, NFAT5 could play a role in permanent extracellular matrix protein production by fibroblasts [34] and could serve as a binding site for glucocorticoid receptor (GR), possibly explaining its anti-proliferative role in fibrosis formation [35]. In healthy myoblasts, NFAT5 is an essential protein in cell migration during myogenesis, but in inflammatory disease pro-inflammatory cytokines hamper normal NFAT5 physiology [36][37]. In lymphocytes and renal medullary cells, hyperosmotic stimuli activate the guanine nucleotide exchange factor Brx, also named A-kinase anchor protein 13 (AKAP13), which belongs to the protein kinase A (PKA) family and is linked with Rac1. In turn, it activates p38αMAPK, glycogen synthase kinase 3 (GSK-3), and NFAT5 [38][39]. The pathway also involves osmolytes, which are protective solutes that safeguard cells from perturbations in volume and osmotic imbalance. Osmolytes are involved in normal skeletal muscle physiology and become dysregulated in DMD (Table 2). Dystrophin deficiency perturbs the muscle cell’s osmotic balance, probably due mostly to the passive efflux of osmolytes through the leaky plasma membranes. Activation of the osmolyte pathway in DMD may act to stabilize proteins and counteract tissue injury.
Table 2. The role of organic osmolytes in skeletal muscle physiology and in Duchenne muscular dystrophy (DMD) and its mouse model mdx.
| Organic Osmolyte | Physiological Role | Expression in DMD Patients/mdx Mouse | Clinical Trials |
|---|---|---|---|
| Taurine | Osmoregulation, antioxidant, modulation of Ca2+ signaling, conjugation of bile acids |
DMD: ↑ taurine excretion ↑ taurine trans-porter (TauT) mdx: ↓ muscle taurine ↓ TauT |
Peripartum cardiomyopathy, thalassemia major, diabetes, etc. |
| Betaine | Osmoregulation, methyldonor |
DMD: expression in small regenerative and atrophic fibers | Homocystinuria, non-alcoholic fatty liver disease, etc. |
| Myo-inositol (MI) | Osmoregulation, structural base for 2nd messengers (signal transduction, component of plasma membrane) | DMD: ↑ sodium/myo-inositol transporter (SMIT) | Polycystic ovary syndrome, bipolar disorders, etc. |
| Sorbitol | Osmoregulation | DMD: ↑ Aldose reductase mdx: ↑ Aldose reductase (cardiac muscle) |
In turn, PKA activates both GSK3 and p38MAPK. The latter has an influence on NFAT5. Upon translocation to the nucleus, NFAT5 has the ability to activate genes coding for organic osmolytes carriers. A graphical representation of this overview is shown in Figure 1.
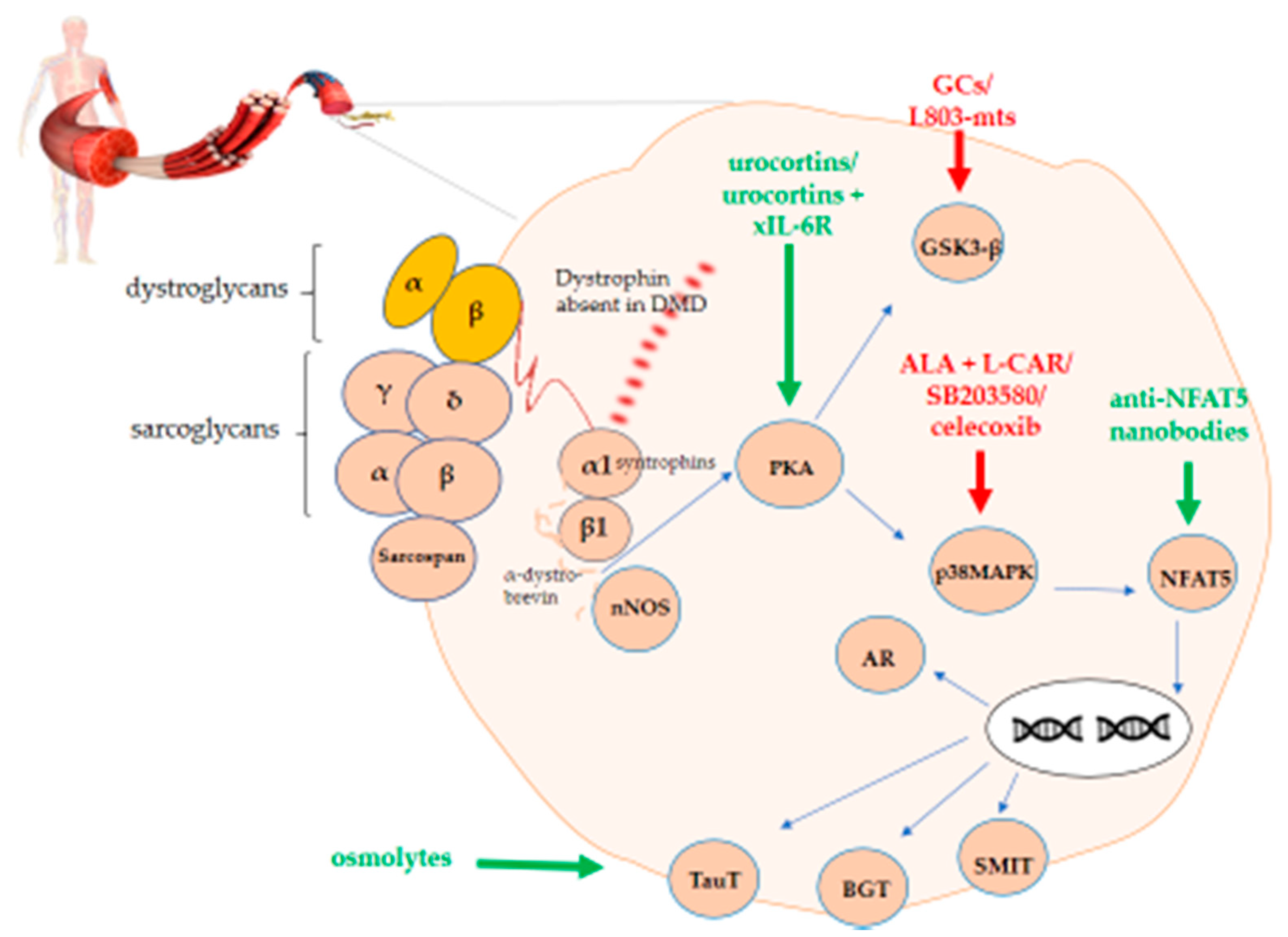
Figure 1. PKA-p38MAPK-NFAT5-organic osmolytes pathway in Duchenne muscular dystrophy (DMD). The figure shows the schematic representation of the protein kinase A/mitogen-activated protein kinase/nuclear factor of activated T-cells 5 (PKA-p38MAPK-NFAT5)-organic osmolytes pathway members representing potential therapeutic targets either to stimulate (green) or to inhibit (red) for treating DMD.
2. The PKA-p38MAPK-NFAT5- Pathway as a Therapeutic Target in Duchenne Muscular Dystrophy
2.1. Protein Kinase A
PKA regulates sugar, glycogen and lipid metabolism in the cell. In skeletal muscle tissue, PKA is in close contact with the DAPC, more specifically with α- and β- dystrobrevin [40]. In mdx mice and DMD patients, PKA is downregulated along with the muscle-specific A-kinase anchor protein (AKAP) named myospryn. PKA activity is reduced by 50% in mdx mice [41].
PKA upregulating molecules, urocortins (Ucns), have been successfully used in mdx mice. Ucns are neuropeptides involved in inflammatory responses, anxiety and renal physiology [42]. In mdx, Ucns were administered daily s.c. for two- to three-week-old mdx mice, resulting in improved muscle resistance to mechanical stress, increased skeletal muscle mass, reduced necrosis in the diaphragm and slow- and fast-twitch muscles, along with normalized calcium influx [43]. When combined with anti-interleukin-6 receptor antibody (xIL-6R), Ucns improved the impaired force in mdx diaphragms, along with muscle shortening and mechanical work production [44]. This combination also resulted in the recovery of respiratory function and pharyngeal dilator muscle force in mdx mice [45][46]. Both molecules have not been tested yet in DMD patients.
2.2. P38 Mitogen-Activated Protein Kinase
P38 MAPK is responsive to cytokines, UV-light, osmotic perturbations and heat shock and plays an important role in apoptosis. In skeletal muscle, constitutive p38 MAPK activation in satellite cells, as seen during ageing, impairs muscle regeneration [47][48]. Very recently, p38 MAPK has been shown to induce cell fusion in myotube formation. Besides, in aging and chronic inflammation, excessive p38 MAPK activation could disrupt skeletal muscle homeostasis, leading to muscle pathology and atrophy [49]. The exact role of p38 MAPK in mdx and DMD is not clear, but a protective role for p38 MAPK signaling is suggested [50]. In mdx mice, p38 MAPK is phosphorylated (p-p38) [51][52][53][54]. Two studies describe a decrease in p-p38 compared to wild type mice at rest [51][54], whereas one study mentions increased phosphorylation in mdx mice compared to wild type mice at rest [53]. The levels of p-p38 increase after treadmill exercise in mdx mice compared to wild type [52]. In one DMD patient, p-p38 was increased compared to normal individuals [55].
p38 MAPK-targeted therapeutic agents have been used in mdx mice with good results: free radical scavenger α-lipoic acid (ALA)/L-carnitine (L-CAR), p38 inhibitor SB203580 and the cyclooxygenase (COX)-2 inhibitor celecoxib [51][53][56]. Carnitine is a fundamental source of acetyl groups. As it acts by transporting long-chain fatty acids into the mitochondrial matrix, its bioavailability is directly related to the rate of ß-oxidation, which was found to be slow in DMD muscle [57], as were reduced carnitine levels [58]. With 95% of carnitine residing in skeletal muscle and since this tissue largely depends on fatty acids as an energy source, its use has been tried in DMD (50mg/kg/day) in an unsuccessful trial [59]. In Becker muscular dystrophy (BMD), a low level of carnitine was found and supplementation has been observed to be beneficial in some cases [60]. Carnitine might be helpful for cardiomyopathy but trials need more strict correlations and biomarkers. The use of steroid, carnitine and branched chain amino acids in DMD has been reviewed in 2015 [61]. ALA/L-CAR induced a reduction in the level of p-p38 in mdx diaphragms [51] and SB203580 increased the survival of mdx myofibers exposed to oxidative stress [53][56]. A four weeks, treatment of six-week-old mdx mice with celecoxib resulted in a fiber type switch from fast to slow phenotype and enhanced muscle fiber integrity and muscle strength. Increased utrophin A expression was noticed in the diaphragm, heart and tibialis anterior [56]. In mdx mice, utrophin reduces the dystrophic phenotype [62]. These three molecules have not yet been investigated in clinical trials involving DMD patients.
2.3. Glycogen Synthase Kinase 3 Beta
The serine/threonine protein kinase glycogen synthase kinase 3 (GSK-3) is ubiquitously expressed and exists in two isoforms, α and β. GSK-3β is a key regulator of a wide range of cellular functions, with a role in neuronal cell development and energy metabolism [63]. In skeletal muscle, it is involved in muscle regeneration [64][65], and its ablation results in accelerated regeneration after atrophy is induced by disuse [66]. Both in mdx mice and in a canine DMD model, the GSK-3 levels are elevated [67][68][69]. The exact role played by GSK-3 in DMD has not been fully investigated [69], but GCs are believed to interact with GSK-3β [70].
The selective GSK-3 peptide inhibitor, L803-mts, increased glucose transporter type 4 (GLUT4) expression in murine muscle tissue; the transporter is altered in DMD and seems to be involved in DMD insulin resistance [71][72]. No further data on GSK-3-targetted treatment is currently available for DMD.
2.4. Nuclear Factor of Activated T-Cells 5
NFAT5 or tonicity-responsive enhancer binding protein (TonEBP) is strongly present in DMD skeletal muscle tissue in nuclei of small myofibers and fibers with central nuclei [55], which could be linked to its role in muscle regeneration [36]. Staining with a Ser 1197-phosphorylated NFAT5 antibody shows this phosphorylation occurs around myonuclei, on the cell membrane and throughout the sarcoplasm in DMD tissue, especially in small fibers. In vascular smooth muscle cells, this phosphorylation appears to prevent NFAT5 from translocating to the nucleus and accumulates in the cytoplasm [73]. NFAT5 nuclear translocation is important during myogenesis [36]. This absence of NFAT5 translocation was observed in normal myoblasts exposed to pro-inflammatory cytokines, where NFAT5 formed aggregates in the sarcoplasm, without showing increased expression [37], and may also be present in DMD fibers, which are chronically exposed to inflammatory cytokines. This could lead to the decreased total NFAT5 protein expression in DMD patients we observe using immunoblotting. In the 15 muscle samples tested, we found constitutive expression in all 11 healthy controls but only in one of the four DMD samples a prominent band was present (Figure 2). NFAT5 has not been investigated in mdx mice so far. When proteins form pathogenic aggregates in the cytoplasm, one therapeutic option that could be considered are protein specific nanobodies [74], which will be discussed further on.

Figure 2. Nuclear factor of activated T-cells 5 (NFAT5) protein expression in skeletal muscle biopsies. Western blots are shown for skeletal biopsies from four patients affected with Duchenne muscular dystrophy (DMD) and from one representative healthy control (C). Total protein extracts were transferred to nitrocellulose membranes following electroblotting. Overnight incubation with 2 µg/mL mouse monoclonal IgG2a anti-NFAT5 (F-9, Santa Cruz) and 0.4 µg/mL anti-GAPDH (Sigma-Aldrich, St. Louis, MO, USA) (the latter used to correct for protein concentration differences between samples) was followed by immunoreaction detection with chemiluminescence (WesternBright™ Sirius, Advansta, Menlo Park, CA, USA) and Proxima 2650 (Isogen Life Science, De Meern, The Netherlands).
2.5. Factors Downstream of Nuclear Factor of Activated T-Cells 5
NFAT5 is known for activating genes that code for TNF-α, heat shock protein 70 (hsp70), and members of osmolyte pathways, in response to hypertonic stimuli [32][75][76]. Under hypertonic conditions, binding of NFAT5 to tonicity-responsive enhancer sequences located at the promotor region results in the increased gene transcription of osmolyte pathway members. In the next sections, individual osmolyte pathway members and their corresponding osmolytes are discussed in detail [77][78][79][80][81][82][83][84][85][86][87][88][89][90][91].
2.5.1. Taurine Transporter
The taurine transporter (TauT or SLC6A6) is principally located on the cell membrane and transports taurine or 2-aminoethyl sulfonic acid from the extracellular environment into the cell. Taurine is an organic osmolyte and functions as an osmoregulator, but also plays a role in calcium homeostasis and exerts anti-inflammatory and antioxidant actions [92][93][94][95][96][97]. In DMD patients, protein levels of TauT are increased, particularly in the CD56+ small regenerating muscle fibers [55], whereas changes in taurine plasma and urinary levels, indicative of increased taurine excretion, have been reported [96][97]. In mdx mice, hampered regulation of the taurine pathway is manifested by a significant decrease of both taurine and its transporter in muscle tissue.
As a possible correlation exists between low taurine levels and muscle impairment [95][98][99][100][101][102], increasing intramuscular taurine levels could improve the mdx disease phenotype. Indeed, studies have found that taurine treatment in mdx mice improved histopathological features such as necrosis [102][103][104] and reduced markers of inflammation and oxidative damage [104][105][106]. Interestingly, Barker et al. reported the favorable effects of prenatal taurine administration (2.5% w/v) in young mdx mice (aged four weeks), whereas the beneficial effect of taurine on histopathological level was almost completely abolished in mice sacrificed at 10 weeks. While intramuscular taurine levels were increased (+25%) upon treatment in four-week-old mice in comparison to untreated mdx mice, the taurine levels were decreased (−22%) in adult treated mdx mice [107]. High taurine supplementation could induce a reduction in both the expression and activity of its transporter [94][108][109][110], which might explain lower intramuscular taurine levels. In the study by Barker et al., however, the protein expression of TauT was reported unchanged [107]. Taurine supplements might also exert beneficial effects on muscle strength. An increase in normalized fore limb muscle force measured by grip strength has been reported [103][111], but the in vitro measurement of specific muscle force was only able to show ameliorated muscle strength in young mdx mice [107], and not in adult mice [103][107]. Furthermore, a synergistic effect of taurine (1 g/kg) and α-methylprednisolone (1 mg/kg) on fore limb strength has been observed in mdx mice [112].
Taurine was able to improve histopathological features and muscle strength in the mdx mouse model and thus makes an interesting compound for the treatment of DMD. To our knowledge, no clinical trials have been conducted to evaluate the effect of taurine in DMD patients; however, the use of taurine in other conditions such as peripartum cardiomyopathy, obesity, etc. has been investigated (https://www.clinicaltrials.gov/, accessed on 10 February 2021).
2.5.2. Betaine Gamma Aminobutyric Acid Transporter
The betaine gamma aminobutyric acid transporter (BGT or SLC6A12) transports betaine and to a lesser extent gamma aminobutyric acid (GABA) into cells. Betaine is an osmolyte that counters osmotic stress, and in addition acts as a methyl group donor in the conversion of homocysteine to dimethylglycine and methionine in the mitochondria of liver and kidney cells [80][81][82][83][93]. To our knowledge, betaine levels in the muscles of DMD patients have not been investigated; however, the increased expression of BGT in the small regenerating and atrophic muscle fibers of DMD patients has been reported, while staining was absent in control muscle tissues [93].
Betaine influences proliferation and differentiation in C2C12 cells [113][114]. The effect of betaine on proliferation and the differentiation in C2C12 myoblasts is abolished in the presence of 5-aza-2 deoxycytidine, a methylation inhibitor, which corroborates the hypothesis that betaine could regulate genes involved in skeletal muscle formation via epigenetic DNA methylation [113]. Furthermore, betaine might alter the distribution between fast- and slow-twitch muscle fibers in favor of slow-twitch myofibers. Slow-twitch fibers are more fatigue-resistant and contain more mitochondria, required for aerobic respiration. Betaine was reported to induce an upregulation of slow-twitch genes [113], presumably by the induction of NFATc1 and the subsequent attenuation of MyoD transcription. On the contrary, transcription of MyoD1 is stimulated in the breast muscle of partridge shank broiler chickens when supplemented with betaine [115]. Dose-dependent effects of betaine and/or differences in the metabolism between species might explain these conflicting results of betaine supplementation on MyoD transcription. In addition, betaine might exert beneficial effects on fatty tissue deposition. Betaine supplementation in mice significantly decreased the percentage of fat mass, whereas lean body mass of mice was increased [113]. Similar results were obtained in a mouse model for obesity. Betaine was able to reduce whole body fat, specifically perirenal, inguinal and gonadal fat, together with inflammatory stress-related genes such as IL-1, IL-6, CXCL13, etc. [116]. Betaine might affect the fatty acid synthesis pathway in muscle and was able to prevent a build-up of intramyocellular lipids. However, Wu et al. described increased lipid accumulation in vitro upon betaine supplementation [117]. These conflicting results might be attributed to differences between in vitro and in vivo models. To note, higher betaine intake in humans is associated with lower body fat [118][119].
Besides a reduction in fat mass, some researchers have reported increased skeletal muscle mass and endurance, both in mice and in humans, upon betaine supplementation [113][118][119][120]. Other beneficial effects of betaine supplementation on skeletal muscle have also been reported. For example, betaine exerted protective effects against skeletal muscle apoptosis caused by chronic alcohol overconsumption. Betaine treatment was able to significantly reduce cytochrome C-release, calpain activity, TNF-α, NF-ƘB, and creatine kinase levels [121]. Betaine supplementation might affect inflammation, skeletal muscle differentiation and lipid accumulation and it could therefore be worthwhile to explore using it as a supportive treatment option in DMD. Betaine is already approved by the Food and Drug Administration (FDA) as an orphan drug for the treatment of homocystinuria but has not yet been tried in DMD.
2.5.3. Sodium/Myo-Inositol Co-transporter
The sodium/myo-inositol co-transporter (SLC5A3 or SMIT) transports myo-inositol (MI) into the cell. MI is a cyclic polyol and exerts numerous physiological functions. Besides its function as an osmolyte, protecting the cell against osmotic stress, MI can be phosphorylated to phosphatidylinositol 4,5-biphosphate (PIP2). PIP2 is a component of the plasma membrane and plays an important role in signal transduction. PIP2 can be further metabolized into diacylglycerol (DAG) or in inositol-(1,4,5) trisphosphate (IP3) that triggers the release of Ca2+ [122][123]. Protein levels of SMIT are readily detected in muscle tissue of DMD patients, in contrast to healthy subjects. Furthermore, immunofluorescent staining in muscle fibers of DMD patients revealed the presence of SMIT in a large part of muscle fibers and inflammatory cells (e.g., CD68+ macrophages and, to a lesser extent, CD206+ macrophages and T cells) [55][93].
The therapeutic effects of MI have been investigated primarily in the context of diabetes mellitus, a disease characterized by high glucose blood levels. Supplementation with MI in glucose-loaded mice caused a decrease in plasma glucose levels [124]. Similar beneficial effects of MI supplementation in obese insulin-resistant Rhesus monkeys on plasma glucose levels have been observed [125]. An MI-induced plasma glucose uptake in myotubes could, however, not be shown in vitro [126], which led to the hypothesis that the effect requires conversion of MI to D-chiro-inositol [124]. Rodriguez et al. reported hyperinsulinemia and insulin resistance in, respectively, 48.5% and 36% of DMD/Becker patients [72]. MI supplements have, however, not yet been tried in DMD patients.
2.5.4. Aldose Reductase
Aldose reductase is an enzyme that catalyzes the reduction of glucose to sorbitol, the first reaction of the polyol pathway. Later, it was shown that its specificity was not limited to glucose and that the enzyme has a wide range preference for hydrophobic substrates such as steroids and lipid-derived/hydrophobic aldehydes [127][128][129][130][131][132][133]. Aldose reductase expression is high in skeletal muscle [134] and is increased even further in the muscle of DMD patients [55][93]. Similarly, an upregulation of aldose reductase is observed in the cardiac tissue of ageing mdx mice [135].
The role of sorbitol and aldose reductase is most extensively studied in models for diabetes. Elevation of sorbitol by 90% has been reported in the skeletal muscle of diabetic rats [136]. Supposedly, a decrease in the availability of adenosine triphosphate (ATP) molecules related to increased polyol pathway activity in diabetes might be associated with muscle dysfunction. Treatment with an aldose reductase inhibitor restored sorbitol levels to normal and had beneficial effects on relaxation and to a lesser extent on the contraction of skeletal muscle [136]. Contrastingly, insulin treatment can prevent rising sorbitol levels in diabetic rats but is incapable of ameliorating muscle strength in a significant manner [137][138]. Thus, the precise role of sorbitol on skeletal muscle dysfunction in diabetes is not entirely clear.
Furthermore, aldose reductase inhibition is also examined as a treatment for diabetes-induced vascular inflammation. Under hyperglycaemic conditions, NF-ƘB activity is enhanced in cultured vascular smooth muscle cells (VSMC) of rats. The research of Ramana et al. demonstrated that sorbinil, an aldose reductase inhibitor, abrogates high glucose mediated NF-ƘB activity presumably by interfering with the phosphorylation of IƘB-α and subsequent inhibition of NF-ƘB translocation to the nucleus. As expected, sorbinil prevents the expression of vascular cell adhesion molecule 1 (VCAM-1) and intracellular adhesion molecule 1 (ICAM-1), both target genes of NF-ƘB, in high glucose cultured VSMC’s. Interestingly, sorbinil is unable to inhibit osmotic stress-induced NF-ƘB activity. Apparently, hyperglycaemic and hyperosmotic conditions mediate NF-ƘB activity in, respectively, a protein kinase C (PKC)-dependent and PKC-independent manner. This difference in regulation might explain why aldose reductase inhibitors are able to inhibit high glucose-induced NF-ƘB activity, but not upon osmotic stress. Studies have reported an anti-inflammatory effect of aldose reductase inhibitors, which could be achieved by the modulation of NF-ƘB activity [139][140][141].
On the other hand, inhibition of aldose reductase aggravates tissue damage in an in vivo temporal artery severe combined immunodeficiency (SCID) mouse model for vasculitis. Aldose reductase plays a role in the detoxification of 4-hydroxynonenal (HNE), a biomarker for lipid peroxidation. Rittner et al. further investigated the relationship between aldose reductase and HNE in vasculitis. Sorbinil treatment in a mouse model for giant cell arteritis induced a twofold and threefold increase in, respectively, HNE adducts and apoptotic cells. Furthermore, stimulation of mononuclear cells with HNE induces the expression of aldose reductase. The latter might explain why an upregulation of aldose reductase is observed in T-cells, macrophages and smooth muscular cells surrounding arteritic lesions together with the presence of HNE in vivo [132]. These observations led to the hypothesis that aldose reductase interferes with lipid peroxidation and protects the arterial wall tissue against oxidative damage. Of note, clinical trials have been conducted to evaluate the effect of sorbinil for the treatment of diabetic retinopathy.
References
- Koenig, M.; Hoffman, E.; Bertelson, C.; Monaco, A.; Feener, C.; Kunkel, L. Complete cloning of the duchenne muscular dystrophy (DMD) cDNA and preliminary genomic organization of the DMD gene in normal and affected individuals. Cell 1987, 50, 509–517.
- Hoffman, E.P.; Brown, R.H.; Kunkel, L.M. Dystrophin: The protein product of the duchenne muscular dystrophy locus. Cell 1987, 51, 919–928.
- Ehmsen, J.; Poon, E.; Davies, K. The dystrophin-associated protein complex. J. Cell Sci. 2002, 115, 2801–2803.
- Rahimov, F.; Kunkel, L.M. Cellular and molecular mechanisms underlying muscular dystrophy. J. Cell Biol. 2013, 201, 499–510.
- Constantin, B. Dystrophin complex functions as a scaffold for signalling proteins. Biochim. Biophys. Acta (BBA) Biomembr. 2014, 1838, 635–642.
- Johnson, E.K.; Li, B.; Yoon, J.H.; Flanigan, K.M.; Martin, P.T.; Ervasti, J.; Montanaro, F. Identification of New Dystroglycan Complexes in Skeletal Muscle. PLoS ONE 2013, 8, e73224.
- Li, D.; Long, C.; Yue, Y.; Duan, D. Sub-physiological sarcoglycan expression contributes to compensatory muscle protection in mdx mice. Hum. Mol. Genet. 2009, 18, 1209–1220.
- Gibbs, E.M.; McCourt, J.L.; Shin, K.M.; Hammond, K.G.; Marshall, J.L.; Crosbie, R.H. Loss of sarcospan exacerbates pathology in mdx mice, but does not affect utrophin amelioration of disease. Hum. Mol. Genet. 2021, 264.
- Peter, A.K.; Marshall, J.L.; Crosbie, R.H. Sarcospan reduces dystrophic pathology: Stabilization of the utrophin–glycoprotein complex. J. Cell Biol. 2008, 183, 419–427.
- Marshall, J.L.; Crosbie-Watson, R.H. Sarcospan: A small protein with large potential for Duchenne muscular dystrophy. Skelet. Muscle 2013, 3, 1.
- Koo, T.; Malerba, A.; Athanasopoulos, T.; Trollet, C.; Boldrin, L.; Ferry, A.; Popplewell, L.; Foster, H.; Foster, K.; Dickson, G. Delivery of AAV2/9-Microdystrophin Genes Incorporating Helix 1 of the Coiled-Coil Motif in the C-Terminal Domain of Dystrophin Improves Muscle Pathology and Restores the Level of α1-Syntrophin and α-Dystrobrevin in Skeletal Muscles of mdx Mice. Hum. Gene Ther. 2011, 22, 1379–1388.
- De Arcangelis, V.; Serra, F.; Cogoni, C.; Vivarelli, E.; Monaco, L.; Naro, F. β1-Syntrophin Modulation by miR-222 in mdx Mice. PLoS ONE 2010, 5, e12098.
- Yoon, J.H.; Johnson, E.; Xu, R.; Martin, L.T.; Martin, P.T.; Montanaro, F. Comparative Proteomic Profiling of Dystroglycan-Associated Proteins in Wild Type, mdx, and Galgt2 Transgenic Mouse Skeletal Muscle. J. Proteome Res. 2012, 11, 4413–4424.
- Peters, M.F.; Sadoulet-Puccio, H.M.; Grady, R.M.; Kramarcy, N.R.; Kunkel, L.M.; Sanes, J.R.; Sealock, R.; Froehner, S.C. Differential Membrane Localization and Intermolecular Associations of α-Dystrobrevin Isoforms in Skeletal Muscle. J. Cell Biol. 1998, 142, 1269–1278.
- Pereira, C.C.D.S.; Kiyomoto, B.H.; Cardoso, R.; Oliveira, A.S.B. Duchenne muscular dystrophy: Alpha-dystroglycan immunoexpression in skeletal muscle and cognitive performance. Arq. Neuro-Psiquiatria 2005, 63, 984–989.
- Wakayama, Y.; Inoue, M.; Kojima, H.; Jimi, T.; Yamashita, S.; Kumagai, T.; Shibuya, S.; Hara, H.; Oniki, H. Altered alpha1-syntrophin expression in myofibers with Duchenne and Fukuyama muscular dystrophies. Histol. Histopathol. 2006, 21, 23–34.
- Cirak, S.; Arechavala-Gomeza, V.; Guglieri, M.; Feng, L.; Torelli, S.; Anthony, K.; Abbs, S.; Garralda, M.E.; Bourke, J.; Wells, D.J.; et al. Exon skipping and dystrophin restoration in patients with Duchenne muscular dystrophy after systemic phosphorodiamidate morpholino oligomer treatment: An open-label, phase 2, dose-escalation study. Lancet 2011, 378, 595–605.
- Compton, A.G.; Cooper, S.T.; Hill, P.M.; Yang, N.; Froehner, S.C.; North, K.N. The Syntrophin-Dystrobrevin Subcomplex in Human Neuromuscular Disorders. J. Neuropathol. Exp. Neurol. 2005, 64, 350–361.
- Metzinger, L.; Blake, D.J.; Squier, M.V.; Anderson, L.V.B.; Deconinck, A.E.; Nawrotzki, R.; Hilton-Jones, D.; Davies, K.E. Dystrobrevin deficiency at the sarcolemma of patients with muscular dystrophy. Hum. Mol. Genet. 1997, 6, 1185–1191.
- Dombernowsky, N.W.; Ölmestig, J.N.; Witting, N.; Kruuse, C. Role of neuronal nitric oxide synthase (nNOS) in Duchenne and Becker muscular dystrophies—Still a possible treatment modality? Neuromuscul. Disord. 2018, 28, 914–926.
- E Brenman, J.; Chao, D.S.; Xia, H.; Aldape, K.; Bredt, D.S. Nitric oxide synthase complexed with dystrophin and absent from skeletal muscle sarcolemma in Duchenne muscular dystrophy. Cell 1995, 82, 743–752.
- Chang, W.J.; Iannaccone, S.T.; Lau, K.S.; Masters, B.S.; McCabe, T.J.; McMillan, K.; Padre, R.C.; Spencer, M.J.; Tidball, J.G.; Stull, J.T. Neuronal nitric oxide synthase and dystrophin-deficient muscular dystrophy. Proc. Natl. Acad. Sci. USA 1996, 93, 9142–9147.
- Crosbie, R.H.; Straub, V.; Yun, H.-Y.; Lee, J.C.; Rafael, J.A.; Chamberlain, J.S.; Dawson, V.L.; Dawson, T.M.; Campbell, K.P. mdx muscle pathology is independent of nNOS perturbation. Hum. Mol. Genet. 1998, 7, 823–829.
- Wehling, M.; Spencer, M.J.; Tidball, J.G. A nitric oxide synthase transgene ameliorates muscular dystrophy in mdx mice. J. Cell Biol. 2001, 155, 123–132.
- Chao, D.S.; Silvagno, F.; Bredt, D.S. Muscular dystrophy in mdx mice despite lack of neuronal nitric oxide synthase. J. Neurochem. 2002, 71, 784–789.
- Rando, T.A. The dystrophin-glycoprotein complex, cellular signaling, and the regulation of cell survival in the muscular dystrophies. Muscle Nerve 2001, 24, 1575–1594.
- Sun, B.; Vaughan, D.M.; Tikunova, S.B.; Creamer, T.P.; Davis, J.P.; Kekenes-Huskey, P.M. Calmodulin–Calcineurin Interaction beyond the Calmodulin-Binding Region Contributes to Calcineurin Activation. Biochemistry 2019, 58, 4070–4085.
- Herbelet, S.; Rodenbach, A.; De Paepe, B.; De Bleecker, J.L. Anti-Inflammatory and General Glucocorticoid Physiology in Skeletal Muscles Affected by Duchenne Muscular Dystrophy: Exploration of Steroid-Sparing Agents. Int. J. Mol. Sci. 2020, 21, 4596.
- Bhatnagar, S.; Kumar, A. Therapeutic targeting of signaling pathways in muscular dystrophy. J. Mol. Med. 2010, 88, 155–166.
- Adcock, I. Glucocorticoid-regulated Transcription Factors. Pulm. Pharmacol. Ther. 2001, 14, 211–219.
- Angelini, C.; Peterle, E. Old and new therapeutic developments in steroid treatment in Duchenne muscular dystrophy. Acta Myol. Myopathies Cardiomyopathies Off. J. Mediterr. Soc. Myol. 2012, 31, 9–15.
- López-Rodríguez, C.; Aramburu, J.; Jin, L.; Rakeman, A.S.; Michino, M.; Rao, A. Bridging the NFAT and NF-κB Families. Immunity 2001, 15, 47–58.
- Choi, S.Y.; Lee-Kwon, W.; Kwon, H.M. The evolving role of TonEBP as an immunometabolic stress protein. Nat. Rev. Nephrol. 2020, 16, 352–364.
- Herbelet, S.; De Paepe, B.; De Bleecker, J.L. Abnormal NFAT5 Physiology in Duchenne Muscular Dystrophy Fibroblasts as a Putative Explanation for the Permanent Fibrosis Formation in Duchenne Muscular Dystrophy. Int. J. Mol. Sci. 2020, 21, 7888.
- Herbelet, S.; De Paepe, B.; De Bleecker, J.L. Description of a Novel Mechanism Possibly Explaining the Antiproliferative Properties of Glucocorticoids in Duchenne Muscular Dystrophy Fibroblasts Based on Glucocorticoid Receptor GR and NFAT5. Int. J. Mol. Sci. 2020, 21, 9225.
- O’Connor, R.S.; Mills, S.T.; Jones, K.A.; Ho, S.N.; Pavlath, G.K. A combinatorial role for NFAT5 in both myoblast migration and differentiation during skeletal muscle myogenesis. J. Cell Sci. 2006, 120, 149–159.
- Herbelet, S.; De Vlieghere, E.; Gonçalves, A.; De Paepe, B.; Schmidt, K.; Nys, E.; Weynants, L.; Weis, J.; Van Peer, G.; Vandesompele, J.; et al. Localization and Expression of Nuclear Factor of Activated T-Cells 5 in Myoblasts Exposed to Pro-inflammatory Cytokines or Hyperosmolar Stress and in Biopsies from Myositis Patients. Front. Physiol. 2018, 9, 126.
- Kino, T.; Takatori, H.; Manoli, I.; Wang, Y.; Tiulpakov, A.; Blackman, M.R.; Su, Y.A.; Chrousos, G.P.; DeCherney, A.H.; Segars, J.H. Brx Mediates the Response of Lymphocytes to Osmotic Stress Through the Activation of NFAT5. Sci. Signal. 2009, 2, ra5.
- Aramburu, J.; López-Rodríguez, C. Regulation of Inflammatory Functions of Macrophages and T Lymphocytes by NFAT5. Front. Immunol. 2019, 10, 535.
- Ceccarini, M.; Grasso, M.; Veroni, C.; Gambara, G.; Artegiani, B.; Macchia, G.; Ramoni, C.; Torreri, P.; Mallozzi, C.; Petrucci, T.C.; et al. Association of Dystrobrevin and Regulatory Subunit of Protein Kinase A: A New Role for Dystrobrevin as a Scaffold for Signaling Proteins. J. Mol. Biol. 2007, 371, 1174–1187.
- Reynolds, J.G.; McCalmon, S.A.; Donaghey, J.A.; Naya, F.J. Deregulated Protein Kinase A Signaling and Myospryn Expression in Muscular Dystrophy. J. Biol. Chem. 2008, 283, 8070–8074.
- Fekete, Éva M.; Zorrilla, E.P. Physiology, pharmacology, and therapeutic relevance of urocortins in mammals: Ancient CRF paralogs. Front. Neuroendocr. 2007, 28, 1–27.
- Reutenauer-Patte, J.; Boittin, F.-X.; Patthey-Vuadens, O.; Ruegg, U.T.; Dorchies, O.M. Urocortins Improve Dystrophic Skeletal Muscle Structure and Function through Both PKA- and Epac-Dependent Pathways. Am. J. Pathol. 2012, 180, 749–762.
- Manning, J.; Buckley, M.M.; O’Halloran, K.D.; O’Malley, D.; Bsc, J.M. Combined XIL-6R and urocortin-2 treatment restores MDX diaphragm muscle force. Muscle Nerve 2017, 56, E134–E140.
- Burns, D.P.; Canavan, L.; Rowland, J.; O’Flaherty, R.; Brannock, M.; Drummond, S.E.; O’Malley, D.; Edge, D.; O’Halloran, K.D. Recovery of respiratory function in mdx mice co-treated with neutralizing interleukin-6 receptor antibodies and urocortin-2. J. Physiol. 2018, 596, 5175–5197.
- Burns, D.P.; Rowland, J.; Canavan, L.; Murphy, K.H.; Brannock, M.; O’Malley, D.; O’Halloran, K.D.; Edge, D. Restoration of pharyngeal dilator muscle force in dystrophin-deficient (mdx) mice following co-treatment with neutralizing interleukin-6 receptor antibodies and urocortin 2. Exp. Physiol. 2017, 102, 1177–1193.
- Cosgrove, B.D.; Gilbert, P.M.; Porpiglia, E.; Mourkioti, F.; Lee, S.P.; Corbel, S.Y.; Llewellyn, M.E.; Delp, S.L.; Blau, H.M. Rejuvenation of the muscle stem cell population restores strength to injured aged muscles. Nat. Med. 2014, 20, 255–264.
- Segalés, J.; Perdiguero, E.; Muñoz-Cánoves, P. Regulation of Muscle Stem Cell Functions: A Focus on the p38 MAPK Signaling Pathway. Front. Cell Dev. Biol. 2016, 4, 91.
- Tomida, T.; Adachi-Akahane, S. Roles of p38 MAPK signaling in the skeletal muscle formation, regeneration, and pathology. Folia Pharmacol. Jpn. 2020, 155, 241–247.
- Shi, H.; Verma, M.; Zhang, L.; Dong, C.; Flavell, R.A.; Bennett, A.M. Improved regenerative myogenesis and muscular dystrophy in mice lacking Mkp5. J. Clin. Investig. 2013, 123, 2064–2077.
- Hnia, K.; Hugon, G.; Rivier, F.; Masmoudi, A.; Mercier, J.; Mornet, D. Modulation of p38 Mitogen-Activated Protein Kinase Cascade and Metalloproteinase Activity in Diaphragm Muscle in Response to Free Radical Scavenger Administration in Dystrophin-Deficient Mdx Mice. Am. J. Pathol. 2007, 170, 633–643.
- Nakamura, A.; Yoshida, K.; Ueda, H.; Takeda, S.; Ikeda, S.-I. Up-regulation of mitogen activated protein kinases in mdx skeletal muscle following chronic treadmill exercise. Biochim. Biophys. Acta (BBA) Mol. Basis Dis. 2005, 1740, 326–331.
- Wissing, E.R.; Boyer, J.G.; Kwong, J.Q.; Sargent, M.A.; Karch, J.; McNally, E.M.; Otsu, K.; Molkentin, J.D. P38α MAPK underlies muscular dystrophy and myofiber death through a Bax-dependent mechanism. Hum. Mol. Genet. 2014, 23, 5452–5463.
- Lang, J.M.; Esser, K.A.; Dupont-Versteegden, E.E. Altered Activity of Signaling Pathways in Diaphragm and Tibialis Anterior Muscle of Dystrophic Mice. Exp. Biol. Med. 2004, 229, 503–511.
- De Paepe, B.; Martin, J.-J.; Herbelet, S.; Jimenez-Mallebrera, C.; Iglesias, E.; Jou, C.; Weis, J.; De Bleecker, J.L. Activation of osmolyte pathways in inflammatory myopathy and Duchenne muscular dystrophy points to osmoregulation as a contributing pathogenic mechanism. Lab. Investig. 2016, 96, 872–884.
- Péladeau, C.; Adam, N.J.; Jasmin, B.J. Celecoxib treatment improves muscle function in mdx mice and increases utrophin A expression. FASEB J. 2018, 32, 5090–5103.
- Carroll, J.E.; Villadiego, A.; Brooke, M.H. Increased long chain acyl CoA in Duchenne muscular dystrophy. Neurology 1983, 33, 1507.
- Berthillier, G.; Eichenberger, D.; Carrier, H.; Guibaud, P.; Got, R. Carnitine metabolism in early stages of Duchenne muscular dystrophy. Clin. Chim. Acta 1982, 122, 369–375.
- Escobar-Cedillo, R.E.; Tintos-Hernandez, J.A.; Martinez-Castro, G.; Montes de Oca-Sanchez, B.; Rodriguez-Jurado, R.; Miranda-Duarte, A. L-carnitine supplementation in duchenne muscular dystrophy steroid-naive patients: A pilot study. Curr. Top. Nutraceut. Res. 2013, 3, 97–102.
- Angelini, C.; Marozzo, R.; Pegoraro, V. Current and emerging therapies in Becker muscular dystrophy (BMD). Acta Myol. Myopathies Cardiomyopathies Off. J. Mediterr. Soc. Myol. 2019, 38, 172–179.
- Angelini, C.; Tasca, E. Drugs in development and dietary approach for Duchenne muscular dystrophy. Orphan Drugs Res. Rev. 2015, 5, 51–60.
- Smythe, G.M.; Forwood, J.K. Altered mitogen-activated protein kinase signaling in dystrophic (mdx) muscle. Muscle Nerve 2012, 46, 374–383.
- E Plyte, S. Glycogen synthase kinase-3: Functions in oncogenesis and development. Biochim. Biophys. Acta (BBA) Bioenerg. 1992, 1114, 147–162.
- Duda, P.; Wiśniewski, J.; Wójtowicz, T.; Wójcicka, O.; Jaśkiewicz, M.; Drulis-Fajdasz, D.; Rakus, D.; McCubrey, J.A.; Gizak, A. Targeting GSK3 signaling as a potential therapy of neurodegenerative diseases and aging. Expert Opin. Ther. Targets 2018, 22, 833–848.
- Kaidanovich-Beilin, O.; Woodgett, J.R. GSK-3: Functional Insights from Cell Biology and Animal Models. Front. Mol. Neurosci. 2011, 4, 40.
- Pansters, N.A.; Schols, A.M.; Verhees, K.J.; De Theije, C.C.; Snepvangers, F.J.; Kelders, M.C.; Ubags, N.D.; Haegens, A.; Langen, R.C. Muscle-specific GSK-3β ablation accelerates regeneration of disuse-atrophied skeletal muscle. Biochim. Biophys. Acta (BBA) Mol. Basis Dis. 2015, 1852, 490–506.
- Villa-Moruzzi, E.; Puntoni, F.; Marin, O. Activation of protein phosphatase-1 isoforms and glycogen synthase kinase-3β in muscle from mdx mice. Int. J. Biochem. Cell Biol. 1996, 28, 13–22.
- Feron, M.; Guevel, L.; Rouger, K.; Dubreil, L.; Arnaud, M.-C.; Ledevin, M.; Megeney, L.A.; Cherel, Y.; Sakanyan, V. PTEN Contributes to Profound PI3K/Akt Signaling Pathway Deregulation in Dystrophin-Deficient Dog Muscle. Am. J. Pathol. 2009, 174, 1459–1470.
- Whitley, K.C.; Hamstra, S.I.; Baranowski, R.W.; Watson, C.J.F.; MacPherson, R.E.K.; MacNeil, A.J.; Roy, B.D.; Vandenboom, R.; Fajardo, V.A. GSK3 inhibition with low dose lithium supplementation augments murine muscle fatigue resistance and specific force production. Physiol. Rep. 2020, 8, e14517.
- Ma, Z.; Zhong, Z.; Zheng, Z.; Shi, X.-M.; Zhang, W. Inhibition of Glycogen Synthase Kinase-3β Attenuates Glucocorticoid-Induced Suppression of Myogenic Differentiation In Vitro. PLoS ONE 2014, 9, e105528.
- Kaidanovich-Beilin, O.; Eldar-Finkelman, H. Long-Term Treatment with Novel Glycogen Synthase Kinase-3 Inhibitor Improves Glucose Homeostasis in ob/ob Mice: Molecular Characterization in Liver and Muscle. J. Pharmacol. Exp. Ther. 2005, 316, 17–24.
- Rodriguezcruz, M.; Sanchez, R.; E Escobar, R.; Cruzguzman, O.D.R.; Lopezalarcon, M.; Garcia, M.B.; Coralvazquez, R.M.; Matute, G.; Wong, A.C.V. Evidence of Insulin Resistance and Other Metabolic Alterations in Boys with Duchenne or Becker Muscular Dystrophy. Int. J. Endocrinol. 2015, 2015, 1–8.
- Scherer, C.; Pfisterer, L.; Wagner, A.H.; Hödebeck, M.; Cattaruzza, M.; Hecker, M.; Korff, T. Arterial Wall Stress Controls NFAT5 Activity in Vascular Smooth Muscle Cells. J. Am. Hear. Assoc. 2014, 3, e000626.
- Jovčevska, I.; Muyldermans, S. The Therapeutic Potential of Nanobodies. BioDrugs 2020, 34, 11–26.
- Neuhofer, W. Role of NFAT5 in Inflammatory Disorders Associated with Osmotic Stress. Curr. Genom. 2010, 11, 584–590.
- Woo, S.K.; Lee, S.D.; Na, K.Y.; Park, W.K.; Kwon, H.M. TonEBP/NFAT5 Stimulates Transcription of HSP70 in Response to Hypertonicity. Mol. Cell. Biol. 2002, 22, 5753–5760.
- Tsai, T.-T.; Danielson, K.G.; Guttapalli, A.; Oguz, E.; Albert, T.J.; Shapiro, I.M.; Risbud, M.V. TonEBP/OREBP Is a Regulator of Nucleus Pulposus Cell Function and Survival in the Intervertebral Disc. J. Biol. Chem. 2006, 281, 25416–25424.
- Ito, T.; Fujio, Y.; Hirata, M.; Takatani, T.; Matsuda, T.; Muraoka, S.; Takahashi, K.; Azuma, J. Expression of taurine transporter is regulated through the TonE (tonicity-responsive element)/TonEBP (TonE-binding protein) pathway and contributes to cytoprotection in HepG2 cells. Biochem. J. 2004, 382, 177–182.
- Yang, Y.J.; Han, Y.Y.; Chen, K.; Zhang, Y.; Liu, X.; Li, S.; Wang, K.Q.; Ge, J.B.; Liu, W.; Zuo, J. TonEBP modulates the protective effect of taurine in ischemia-induced cytotoxicity in cardiomyocytes. Cell Death Dis. 2015, 6, e2025.
- Kempson, S.A.; Zhou, Y.; Danbolt, N.C. The betaine/GABA transporter and betaine: Roles in brain, kidney, and liver. Front. Physiol. 2014, 5, 159.
- Kempson, S.A.; Montrose, M.H. Osmotic regulation of renal betaine transport: Transcription and beyond. Pflüger’s Arch. Gesammte Phys. Menschen Tiere 2004, 449, 227–234.
- Kempson, S.A.; Parikh, V.; Xi, L.; Chu, S.; Montrose, M.H. Subcellular redistribution of the renal betaine transporter during hypertonic stress. Am. J. Physiol. Physiol. 2003, 285, C1091–C1100.
- Kempson, S.A.; Beck, J.A.; Lammers, P.E.; Gens, J.S.; Montrose, M.H. Membrane insertion of betaine/GABA transporter during hypertonic stress correlates with nuclear accumulation of TonEBP. Biochim. Biophys. Acta (BBA) Biomembr. 2005, 1712, 71–80.
- Nakanishi, T.; Turner, R.J.; Burg, M.B. Osmoregulatory changes in myo-inositol transport by renal cells. Proc. Natl. Acad. Sci. USA 1989, 86, 6002–6006.
- Kwon, H.M.; Yamauchi, A.; Uchida, S.; Robey, R.B.; Garcia-Perez, A.; Burg, M.B.; Handler, J.S. Renal Na-myo-inositol cotransporter mRNA expression in Xenopus oocytes: Regulation by hypertonicity. Am. J. Physiol. Physiol. 1991, 260, F258–F263.
- Lee, S.D.; Choi, S.Y.; Lim, S.W.; Lamitina, S.T.; Ho, S.N.; Go, W.Y.; Kwon, H.M. TonEBP stimulates multiple cellular pathways for adaptation to hypertonic stress: Organic osmolyte-dependent and -independent pathways. Am. J. Physiol. Physiol. 2011, 300, F707–F715.
- Rim, J.S.; Atta, M.G.; Dahl, S.C.; Berry, G.T.; Handler, J.S.; Kwon, H.M. Transcription of the Sodium/myo-Inositol Cotransporter Gene Is Regulated by Multiple Tonicity-responsive Enhancers Spread over 50 Kilobase Pairs in the 5′-Flanking Region. J. Biol. Chem. 1998, 273, 20615–20621.
- López-Rodríguez, C.; Antos, C.L.; Shelton, J.M.; Richardson, J.A.; Lin, F.; Novobrantseva, T.I.; Bronson, R.T.; Igarashi, P.; Rao, A.; Olson, E.N. Loss of NFAT5 results in renal atrophy and lack of tonicity-responsive gene expression. Proc. Natl. Acad. Sci. USA 2004, 101, 2392–2397.
- Mak, M.C.; Lam, K.M.; Chan, P.K.; Lau, Y.B.; Tang, W.H.; Yeung, P.K.K.; Ko, B.C.B.; Chung, S.M.S.; Chung, S.K. Embryonic Lethality in Mice Lacking the Nuclear Factor of Activated T Cells 5 Protein Due to Impaired Cardiac Development and Function. PLoS ONE 2011, 6, e19186.
- Buccafusca, R.; Venditti, C.P.; Kenyon, L.C.; Johanson, R.A.; Van Bockstaele, E.; Ren, J.; Pagliardini, S.; Minarcik, J.; Golden, J.A.; Coady, M.J.; et al. Characterization of the null murine sodium/myo-inositol cotransporter 1 (Smit1 or Slc5a3) phenotype: Myo-inositol rescue is independent of expression of its cognate mitochondrial ribosomal protein subunit 6 (Mrps6) gene and of phosphatidylinositol levels in neonatal brain. Mol. Genet. Metab. 2008, 95, 81–95.
- Ko, B.C.B.; Ruepp, B.; Bohren, K.M.; Gabbay, K.H.; Chung, S.S.M. Identification and Characterization of Multiple Osmotic Response Sequences in the Human Aldose Reductase Gene. J. Biol. Chem. 1997, 272, 16431–16437.
- Jhiang, S.M.; Fithian, L.; Smanik, P.; McGill, J.; Tong, Q.; Mazzaferri, E.L. Cloning of the human taurine transporter and characterization of taurine uptake in thyroid cells. FEBS Lett. 1993, 318, 139–144.
- De Paepe, B. Osmolytes as Mediators of the Muscle Tissue’s Responses to Inflammation: Emerging Regulators of Myositis with Therapeutic Potential. Eur. Med. J. Rheumatol. 2017, 4, 83–89.
- Tappaz, M.L. Taurine Biosynthetic Enzymes and Taurine Transporter: Molecular Identification and Regulations. Neurochem. Res. 2004, 29, 83–96.
- Terrill, J.R.; Grounds, M.D.; Arthur, P.G. Taurine deficiency, synthesis and transport in the mdx mouse model for Duchenne Muscular Dystrophy. Int. J. Biochem. Cell Biol. 2015, 66, 141–148.
- Bank, W.J.; Rowland, L.P.; Ipsen, J. Amino Acids of Plasma and Urine in Diseases of Muscle. Arch. Neurol. 1971, 24, 176–186.
- Wen, C.; Li, F.; Zhang, L.; Duan, Y.; Guo, Q.; Wang, W.; He, S.; Li, J.; Yin, Y. Taurine is Involved in Energy Metabolism in Muscles, Adipose Tissue, and the Liver. Mol. Nutr. Food Res. 2019, 63, e1800536.
- Warskulat, U.; Heller-Stilb, B.; Oermann, E.; Zilles, K.; Haas, H.; Lang, F.; Häussinger, D. Phenotype of the Taurine Transporter Knockout Mouse. Methods Enzymol. 2007, 428, 439–458.
- Warskulat, U.; Borsch, E.; Reinehr, R.; Heller-Stilb, B.; Roth, C.; Witt, M.; Häussinger, D. Taurine deficiency and apoptosis: Findings from the taurine transporter knockout mouse. Arch. Biochem. Biophys. 2007, 462, 202–209.
- Ito, T.; Kimura, Y.; Uozumi, Y.; Takai, M.; Muraoka, S.; Matsuda, T.; Ueki, K.; Yoshiyama, M.; Ikawa, M.; Okabe, M.; et al. Taurine depletion caused by knocking out the taurine transporter gene leads to cardiomyopathy with cardiac atrophy. J. Mol. Cell. Cardiol. 2008, 44, 927–937.
- Warskulat, U.; Flögel, U.; Jacoby, C.; Hartwig, H.; Thewissen, M.; Merx, M.W.; Molojavyi, A.; Heller-Stilb, B.; Schrader, J.; Häussinger, D. Taurine transporter knockout depletes muscle taurine levels and results in severe skeletal muscle impairment but leaves cardiac function uncompromised. FASEB J. 2004, 18, 577–579.
- Terrill, J.R.; Webb, S.M.; Arthur, P.G.; Hackett, M.J. Investigation of the effect of taurine supplementation on muscle taurine content in the mdx mouse model of Duchenne muscular dystrophy using chemically specific synchrotron imaging. Analyst 2020, 145, 7242–7251.
- Capogrosso, R.F.; Cozzoli, A.; Mantuano, P.; Camerino, G.M.; Massari, A.M.; Sblendorio, V.T.; De Bellis, M.; Tamma, R.; Giustino, A.; Nico, B.; et al. Assessment of resveratrol, apocynin and taurine on mechanical-metabolic uncoupling and oxidative stress in a mouse model of duchenne muscular dystrophy: A comparison with the gold standard, α-methyl prednisolone. Pharmacol. Res. 2016, 106, 101–113.
- Terrill, J.R.; Grounds, M.D.; Arthur, P.G. [MD-16-0004R1] Increased taurine in pre-weaned juvenile mdx mice greatly reduces the acute onset of myofibre necrosis and dystropathology and prevents inflammation. PLoS Curr. 2016, 8.
- Terrill, J.R.; Pinniger, G.J.; Nair, K.V.; Grounds, M.D.; Arthur, P.G. Beneficial effects of high dose taurine treatment in juvenile dystrophic mdx mice are offset by growth restriction. PLoS ONE 2017, 12, e0187317.
- Terrill, J.R.; Pinniger, G.J.; Graves, J.A.; Grounds, M.D.; Arthur, P.G. Increasing taurine intake and taurine synthesis improves skeletal muscle function in the mdx mouse model for Duchenne muscular dystrophy. J. Physiol. 2016, 594, 3095–3110.
- Murphy, R.M.; Barker, R.G.; Horvath, D.; Van Der Poel, C. Benefits of Pre-natal Taurine Supplementation in Preventing the Onset of Acute Damage in the Mdx Mouse. PLoS Curr. 2017, 9, 9.
- Voss, J.W.; Pedersen, S.F.; Christensen, S.T.; Lambert, I.H. Regulation of the expression and subcellular localization of the taurine transporter TauT in mouse NIH3T3 fibroblasts. JBIC J. Biol. Inorg. Chem. 2004, 271, 4646–4658.
- Bitoun, M.; Tappaz, M. Taurine Down-Regulates Basal and Osmolarity-Induced Gene Expression of Its Transporter, but Not the Gene Expression of Its Biosynthetic Enzymes, in Astrocyte Primary Cultures. J. Neurochem. 2002, 75, 919–924.
- Matsell, D.G.; Bennett, T.; Han, X.; Budreau, A.M.; Chesney, R.W. Regulation of the taurine transporter gene in the S3 segment of the proximal tubule. Kidney Int. 1997, 52, 748–754.
- De Luca, A.; Pierno, S.; Liantonio, A.; Cetrone, M.; Camerino, C.; Fraysse, B.; Mirabella, M.; Servidei, S.; Rüegg, U.T.; Camerino, D.C. Enhanced Dystrophic Progression in mdx Mice by Exercise and Beneficial Effects of Taurine and Insulin-Like Growth Factor-1. J. Pharmacol. Exp. Ther. 2003, 304, 453–463.
- Cozzoli, A.; Rolland, J.-F.; Capogrosso, R.F.; Sblendorio, V.T.; Longo, V.; Simonetti, S.; Nico, B.; De Luca, A. Evaluation of potential synergistic action of a combined treatment with alpha-methyl-prednisolone and taurine on the mdx mouse model of Duchenne muscular dystrophy. Neuropathol. Appl. Neurobiol. 2011, 37, 243–256.
- Du, J.; Shen, L.; Zhang, P.; Tan, Z.; Cheng, X.; Luo, J.; Zhao, X.; Yang, Q.; Gu, H.; Jiang, A.; et al. The regulation of skeletal muscle fiber-type composition by betaine is associated with NFATc1/MyoD. J. Mol. Med. 2018, 96, 685–700.
- Senesi, P.; Luzi, L.; Montesano, A.; Mazzocchi, N.; Terruzzi, I. Betaine supplement enhances skeletal muscle differentiation in murine myoblasts via IGF-1 signaling activation. J. Transl. Med. 2013, 11, 174.
- Chen, R.; Zhuang, S.; Chen, Y.; Cheng, Y.; Wen, C.; Zhou, Y. Betaine improves the growth performance and muscle growth of partridge shank broiler chickens via altering myogenic gene expression and insulin-like growth factor-1 signaling pathway. Poult. Sci. 2018, 97, 4297–4305.
- Du, J.; Shen, L.; Tan, Z.; Zhang, P.; Zhao, X.; Xu, Y.; Gan, M.; Yang, Q.; Ma, J.; Jiang, A.; et al. Betaine Supplementation Enhances Lipid Metabolism and Improves Insulin Resistance in Mice Fed a High-Fat Diet. Nutrients 2018, 10, 131.
- Wu, W.; Wang, S.; Xu, Z.; Wang, X.; Feng, J.; Shan, T.; Wang, Y. Betaine promotes lipid accumulation in adipogenic-differentiated skeletal muscle cells through ERK/PPARγ signalling pathway. Mol. Cell. Biochem. 2018, 447, 137–149.
- Long, J.-A.; Zhong, R.-H.; Chen, S.; Wang, F.; Luo, Y.; Lu, X.-T.; Yishake, D.; Chen, Y.-M.; Fang, A.-P.; Zhu, H.-L. Dietary betaine intake is associated with skeletal muscle mass change over 3 years in middle-aged adults: The Guangzhou Nutrition and Health Study. Br. J. Nutr. 2021, 125, 440–447.
- Gao, X.; Wang, Y.; Randell, E.; Pedram, P.; Yi, Y.; Gulliver, W.; Sun, G. Higher Dietary Choline and Betaine Intakes Are Associated with Better Body Composition in the Adult Population of Newfoundland, Canada. PLoS ONE 2016, 11, e0155403.
- Hoffman, J.R.; A Ratamess, N.; Kang, J.; Rashti, S.L.; Faigenbaum, A.D. Effect of betaine supplementation on power performance and fatigue. J. Int. Soc. Sports Nutr. 2009, 6, 7–10.
- Ilhan, A.O.; Ozkoc, M.; Ol, K.K.; Karimkhani, H.; Senturk, H.; Burukoglu, D.; Kanbak, G. Protective effect of betaine against skeleton muscle apoptosis in rats induced by chronic alcohol and statin consumption. Bratisl. Med. J. 2020, 121, 589–599.
- Berry, G.T.; Mallee, J.J.; Kwon, H.; Rim, J.S.; Mulla, W.R.; Muenke, M.; Spinner, N.B. The human osmoregulatory Na+/myo-inositol cotransporter gene (SLC5A3): Molecular cloning and localization to chromosome 21. Genomics 1995, 25, 507–513.
- Huber, K.; Walk, C.; Kühn, I.; Stein, H.; Kidd, M.; Rodehutscord, M. Chapter 4 Cellular myo-inositol metabolism. In Phytate Destruction—Consequences for Precision Animal Nutrition; Wageningen Academic Publishers: Wageningen, The Netherlands, 2016; pp. 53–60.
- Dang, N.T.; Mukai, R.; Yoshida, K.-I.; Ashida, H. D-Pinitol andmyo-Inositol Stimulate Translocation of Glucose Transporter 4 in Skeletal Muscle of C57BL/6 Mice. Biosci. Biotechnol. Biochem. 2010, 74, 1062–1067.
- Ortmeyer, H.K. Dietary Myoinositol Results in Lower Urine Glucose and in Lower Postprandial Plasma Glucose in Obese Insulin Resistant Rhesus Monkeys. Obes. Res. 1996, 4, 569–575.
- Yap, A.; Nishiumi, S.; Yoshida, K.-I.; Ashida, H. Rat L6 myotubes as an in vitro model system to study GLUT4-dependent glucose uptake stimulated by inositol derivatives. Cytotechnology 2007, 55, 103–108.
- Ramana, K.V.; Srivastava, S.K. Aldose reductase: A novel therapeutic target for inflammatory pathologies. Int. J. Biochem. Cell Biol. 2010, 42, 17–20.
- Petrash, J.M. All in the family: Aldose reductase and closely related aldo-keto reductases. Cell. Mol. Life Sci. 2004, 61, 737–749.
- Morrison, A.D.; Clements, R.S.; Travis, S.B.; Oski, F.; Winegrad, A.I. Glucose utilization by the polyol pathway in human erythrocytes. Biochem. Biophys. Res. Commun. 1970, 40, 199–205.
- González, R.G.; Barnett, P.; Aguayo, J.; Cheng, H.-M.; Chylack, L.T. Direct Measurement of Polyol Pathway Activity in the Ocular Lens. Diabetes 1984, 33, 196–199.
- Yabe-Nishimura, C. Aldose reductase in glucose toxicity: A potential target for the prevention of diabetic complications. Pharmacol. Rev. 1998, 50, 21–34.
- Rittner, H.L.; Hafner, V.; Klimiuk, P.A.; Szweda, L.I.; Goronzy, J.J.; Weyand, C.M. Aldose reductase functions as a detoxification system for lipid peroxidation products in vasculitis. J. Clin. Investig. 1999, 103, 1007–1013.
- Choudhary, S.; Xiao, T.; Srivastava, S.; Zhang, W.; Chan, L.; Vergara, L.; Van Kuijk, F.; Ansari, N. Toxicity and detoxification of lipid-derived aldehydes in cultured retinal pigmented epithelial cells. Toxicol. Appl. Pharmacol. 2005, 204, 122–134.
- O’Connor, T.; Ireland, L.S.; Harrison, D.J.; Hayes, J.D. Major differences exist in the function and tissue-specific expression of human aflatoxin B1 aldehyde reductase and the principal human aldo-keto reductase AKR1 family members. Biochem. J. 1999, 343, 487–504.
- Holland, A.; Dowling, P.; Zweyer, M.; Swandulla, D.; Henry, M.; Clynes, M.; Ohlendieck, K. Proteomic profiling of cardiomyopathic tissue from the aged mdx model of Duchenne muscular dystrophy reveals a drastic decrease in laminin, nidogen and annexin. Proteomics 2013, 13, 2312–2323.
- Cotter, M.; Cameron, N.; Robertson, S.; Ewing, I. Polyol pathway-related skeletal muscle contractile and morphological abnormalities in diabetic rats. Exp. Physiol. 1993, 78, 139–155.
- Sanchez, O.A.; Snow, L.M.; Lowe, D.A.; Serfass, R.C.; Thompson, L.V. Effects of endurance exercise-training on single-fiber contractile properties of insulin-treated streptozotocin-induced diabetic rats. J. Appl. Physiol. 2005, 99, 472–478.
- Sanchez, O.A.; Walseth, T.F.; Snow, L.M.; Serfass, R.C.; Thompson, L.V. Skeletal Muscle Sorbitol Levels in Diabetic Rats with and without Insulin Therapy and Endurance Exercise Training. Exp. Diabetes Res. 2009, 2009, 1–6.
- Ramana, K.V.; Friedrich, B.; Srivastava, S.; Bhatnagar, A. Activation of Nulcear Factor- B by Hyperglycemia in Vascular Smooth Muscle Cells Is Regulated by Aldose Reductase. Diabetes 2004, 53, 2910–2920.
- Ramana, K.V.; Srivastava, S.K. Mediation of aldose reductase in lipopolysaccharide-induced inflammatory signals in mouse peritoneal macrophages. Cytokine 2006, 36, 115–122.
- Reddy, A.B.; Srivastava, S.K.; Ramana, K.V. Anti-inflammatory effect of aldose reductase inhibition in murine polymicrobial sepsis. Cytokine 2009, 48, 170–176.

