1.Duchenne Muscular Dystrophy and PKA-p38MAPK-NFAT5-Pathway
Duchenne muscular dystrophy (DMD) is a severe X-linked disorder usually recognized in early childhood, characterized by progressive skeletal muscle weakness causing loss of ambulation by early adolescence. In addition to the skeletal muscle, the disorder also involves cardiac and respiratory muscles. DMD is caused by disruptive mutations in the
DMD gene, resulting in the absence of its protein product dystrophin from muscle fiber membranes. Contraction damages the dystrophin-deficient muscle fibers, generating cycles of muscle fiber necrosis and regeneration that fail to restore the tissue, leading to fibrosis and fatty replacement. Absence of dystrophin distorts anchoring of the extracellular matrix to the myofiber cytoskeleton [
1,
2,
3]. Dystrophin is part of a larger group of transmembrane proteins, called the dystrophin associated protein complex (DAPC), which has the ability to receive and transduce signals from in- and outside the myofiber and absorb shocks during muscle contraction [
4,
5]. This shock-absorbing effect of the DAPC is compromised in DMD patients and causes cell membrane instability, which start as soon as the fetus can move in utero. The DAPC is composed of dystrobrevins, dystroglycans, sarcoglycans, sarcospan, and syntrophin. The most widely used murine model for DMD are mdx mice that carry a nonsense mutation in exon 23 of the
dystrophin gene. However, the disease phenotype of this murine model is milder than the human condition. While humans display progressive muscle weakness, mice develop active muscle tissue degeneration and regeneration at a young age, which later on continues at a slower pace. The DAPC is affected in DMD and mdx displaying both common and differential deficiencies (). In mdx mice, α- and β-dystroglycans are unstable, whereas expression of α-, β-, γ- and δ-sarcoglycans, sarcospan and α1-syntrophin is weak [
6,
7,
8,
9,
10,
11] and expression of β1-syntrophin and α-dystrobrevin is absent [
12,
13,
14]. In DMD patients, the following DAPC proteins are less expressed to sometimes absent: α- dystroglycan, α-sarcoglycan, sarcospan, α1-syntrophin, α-dystrobrevin, and neuronal nitric oxide synthase (nNOS) [
10,
15,
16,
17,
18,
19,
20,
21,
22]. The role of nNOS instability in mdx pathology is controversial. Indeed, normal nNOS activity reduced dystrophic symptoms in one mdx study, whereas mdx mice crossed with NOS-null mice showed no difference in muscle pathology when compared to mdx mice [
23,
24,
25]. β-dystroglycan is still present in DMD skeletal muscle tissue [
6] and expression studies of β1-syntrophin in DMD patients are not yet available. Both β-dystroglycan and syntrophin function as signaling proteins. More specifically, β-dystroglycan signals to Ras-related C3 botulinum toxin substrate 1 (Rac1) small guanosinetrifosfaat (GTP)ase and to mitogen-activated protein kinase (MAPK) through growth factor receptor-bound protein 2 (Grb2). Syntrophin organizes a signalplex linked to dystrophin and regulates signaling proteins such as voltage-gated sodium channels along with plasma membrane calcium pumps and nNOS. DAPC also interacts with calmodulin which is stimulated by calcium and in turn signals to calcineurin [
5,
26,
27].
Table 1. Differential expression of the main dystrophin-associated protein complex (DAPC) components in Duchenne muscular dystrophy (DMD) and in the mdx mouse.
Hampered signal transduction and cellular pathways have been described in DMD with Toll-like receptor/tumor necrosis factor α (TNF-α)/interleukin 1β (IL-1β)/interleukin 6 (IL-6)-nuclear factor kappa-light-chain-enhancer of the activated B cell pathway (NF-ƘB), Janus kinase/signal transducer and activator of transcription proteins, and the transforming growth factor-β (TGF-β) pathways having been extensively studied and considered for therapeutic targeting [
28,
29]. Due to the important inflammatory aspect of the disease, glucocorticoids (GCs) are the drugs of choice in DMD with their major mode of action residing in the binding to NF-ƘB and MAPKs along with nuclear translocation of nuclear factor of activated T-cells (NFAT) [
30,
31]. The NFAT group consists of five transcription factors belonging to the larger ReI family which also encompasses NF-ƘB. NFATc1-4 are regulated by calcineurin, whereas NFAT5 is a non-calcineurin mediated transcription factor harboring similarities with both NFATc and NF-ƘB [
32]. NFAT5 is a multifaceted protein, which tightly controls cell volume in order to remain inside the homeostatic range. It controls cell growth in embryogenic tissue, mediates inflammation and protects cells from oxidative stress and metabolic aberrations due for instance to excessive caloric intake. It therefore deserves the name of immunometabolic stress protein [
33].
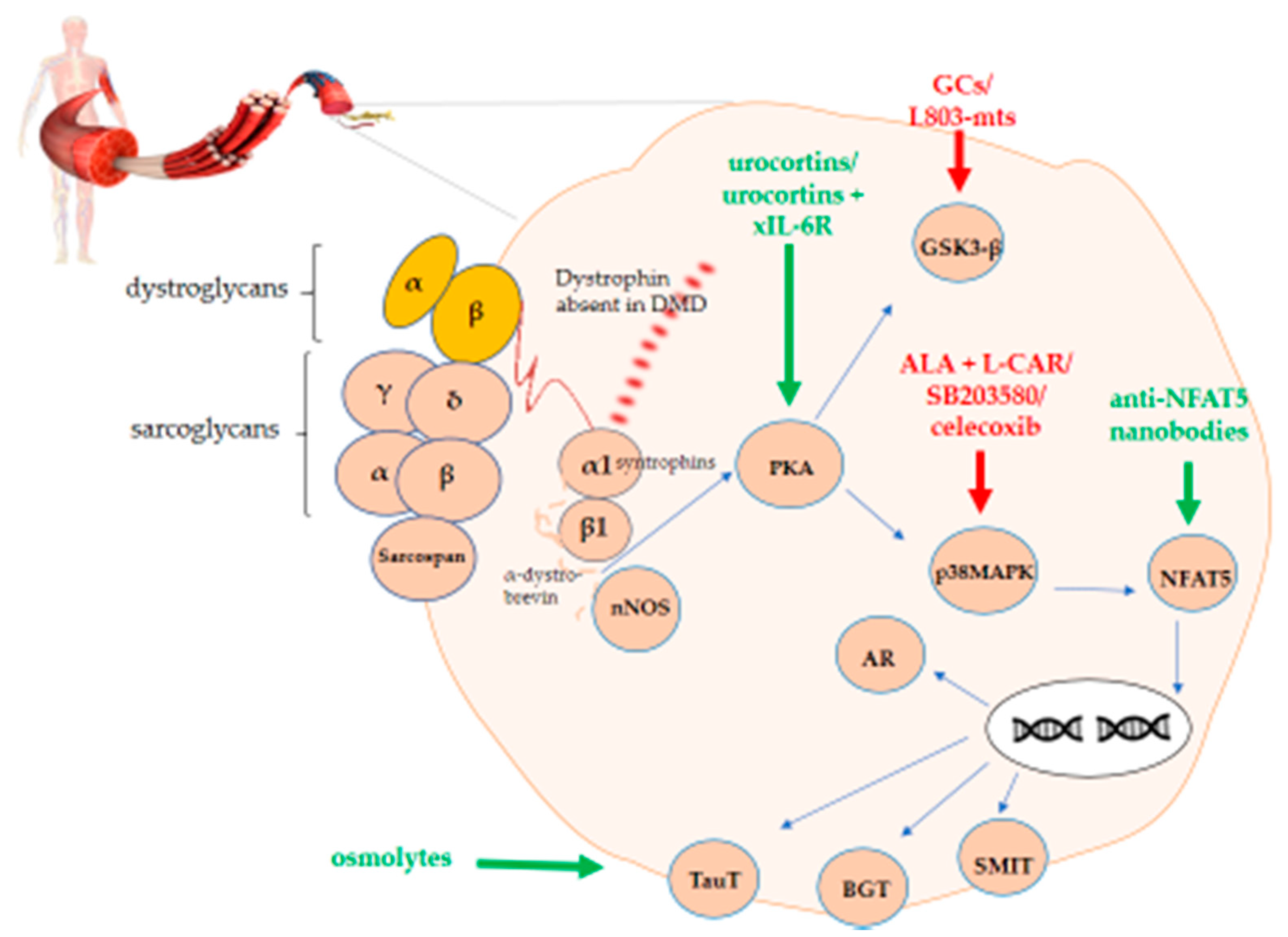
In DMD, NFAT5 could play a role in permanent extracellular matrix protein production by fibroblasts [
34] and could serve as a binding site for glucocorticoid receptor (GR), possibly explaining its anti-proliferative role in fibrosis formation [
35]. In healthy myoblasts, NFAT5 is an essential protein in cell migration during myogenesis, but in inflammatory disease pro-inflammatory cytokines hamper normal NFAT5 physiology [
36,
37]. In lymphocytes and renal medullary cells, hyperosmotic stimuli activate the guanine nucleotide exchange factor Brx, also named A-kinase anchor protein 13 (AKAP13), which belongs to the protein kinase A (PKA) family and is linked with Rac1. In turn, it activates p38αMAPK, glycogen synthase kinase 3 (GSK-3), and NFAT5 [
38,
39]. The pathway also involves osmolytes, which are protective solutes that safeguard cells from perturbations in volume and osmotic imbalance. Osmolytes are involved in normal skeletal muscle physiology and become dysregulated in DMD (). Dystrophin deficiency perturbs the muscle cell’s osmotic balance, probably due mostly to the passive efflux of osmolytes through the leaky plasma membranes. Activation of the osmolyte pathway in DMD may act to stabilize proteins and counteract tissue injury.
Table 2. The role of organic osmolytes in skeletal muscle physiology and in Duchenne muscular dystrophy (DMD) and its mouse model mdx.
In turn, PKA activates both GSK3 and p38MAPK. The latter has an influence on NFAT5. Upon translocation to the nucleus, NFAT5 has the ability to activate genes coding for organic osmolytes carriers. A graphical representation of this overview is shown in .
Figure 1. PKA-p38MAPK-NFAT5-organic osmolytes pathway in Duchenne muscular dystrophy (DMD). The figure shows the schematic representation of the protein kinase A/mitogen-activated protein kinase/nuclear factor of activated T-cells 5 (PKA-p38MAPK-NFAT5)-organic osmolytes pathway members representing potential therapeutic targets either to stimulate (green) or to inhibit (red) for treating DMD.