 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Soraya Smaili | + 1740 word(s) | 1740 | 2021-04-24 14:30:39 | | | |
| 2 | Lindsay Dong | + 252 word(s) | 1992 | 2021-04-26 03:15:08 | | |
Video Upload Options
The family of coronaviruses (CoVs) uses the autophagy machinery of host cells to promote their growth and replication. Pharmacological or pharmacogenomics tools might be used to modulate autofaphy, and these processes stand out potential targets to combat COVID-19.
1. Introduction
On 2 September 2020, WHO recommended corticosteroids as an effective treatment for seriously ill COVID-19 patients. Several other drugs were clinically used in the same effort to contain the deaths caused by COVID-19. Only in December 2020, the Medicines and Healthcare products Regulatory Agency (MHRA) in the United Kingdom and the Food and Drug Administration (FDA) in the United States of America (USA) authorized the emergency use of Pfizer/BioNTech’s and Moderna’s vaccines against COVID-19 [1]. Nevertheless, worldwide vaccine plans are yet to be implemented and novel mutations of the SARS-CoV-2 are rapidly emerging [2][3] demanding continuous research on therapeutics to manage COVID-19.
2. Coronavirus Hijack the Autophagy Machinery to Foster Replication
Autophagy is triggered by the inhibition of mammalian target of rapamycin complex 1 (mTORC1), the primary regulator of nutrient signaling. Moreover, the mTORC1 complex is modulated by upstream regulators that transduce growth factors and energy signals. Autophagosome formation and its self-assembly are coordinated by enzymes and proteins located in the ER, such as phosphatidylinositol 3-phosphate (PI3P) and B-cell lymphoma 2 (BCL-2) interacting proteins Beclin-1/vacuolar protein sorting 34 (Beclin-1/Vps34) complex [4][5]. In addition to these proteins, the activating molecule in Beclin-1-regulated autophagy (Ambra1) plays a vital role as a regulator of autophagy, binding to Beclin-1, promoting the autophagosome formation [6]. Components of the autophagy machinery also participate in the secretion of invading pathogens. The use of autophagic machinery by CoVs was demonstrated, where the initiation of vesicle formation was inhibited by knocking out autophagy-related gene 5 (ATG5) or by wortmannin, suggesting that nsp6-induced autophagy was dependent on Atg5 and PI3K. Finally, transfecting the SARS-CoV open reading frame -8b and -3a into 293T and HeLa cells triggers lysosomal damage and ER stress, consequently inducing the translocation of Transcription Factor EB (TFEB) to the nucleus, a master regulator of lysosomal biogenesis and favoring the transcription of autophagy- and lysosome-related genes [7][8]. Defects in the molecular machinery for macroautophagy, such as the genetic inhibition of ATG5 or beclin-1 (BECN1) genes, consequently make mice and primary human astrocytes more susceptible to viral infections [9][10][11].
Conversely, other studies have highlighted the inhibitory effects of CoV nonstructural proteins on autophagy flux. In fact, overexpressing CoVs membrane-associated papain-like protease PLP2 (PLP2-TM) resulted in inhibition of autophagosome–lysosome fusion and blockade of autophagic flux in HEK293T, HeLa and MCF-7 cells [12]. Likewise, recent evidence described that Vero B4 cells infected with MERS-CoV exhibited reduced Beclin-1 levels, enhanced K48-polyubiquitylation of Beclin-1, reduced Atg14 oligomerization and blocked autophagosome-lysosome fusion [13]. Correspondingly, temporal kinome analysis of Huh7 and MRC5 cells infected with MERS-CoV displayed upregulated PI3K/AKT/mTOR and extracellular signal-regulated kinase/mitogen-activated protein kinase (ERK/MAPK)-mediated signaling [14].
An in-depth analysis of autophagy signaling, and metabolomics corroborated the notion that CoVs modulate PI3K/AKT/mTOR and AMPK signaling, showing that SARS-CoV-2 reduced glycolysis and protein translation by limiting the activation of mTORC1 and AMPK. It was shown that SARS-CoV-2 infection also downregulated spermidine and facilitated AKT1/S-phase kinase-associated protein 2 (SKP2)-dependent degradation of Beclin-1 [15].
Since autophagy may be one of the molecular mechanisms that allow cell invasion and virus replication, it is possible that some mutations may alter the autophagic process [16][17]. SARS-CoV-2, like any type of virus, accumulates mutations over time, and most of these mutations do not implicate in biological effects. However, some key mutations can alter viral biology to the extent of causing changes in its transmission and infection capacity [18]. Briefly, Table 1 summarizes the molecular machinery recruited in autophagy initiation and Figure 1 shows that autophagy mechanisms represent potential targets for pharmacological inhibition of CoVs infection and replication.
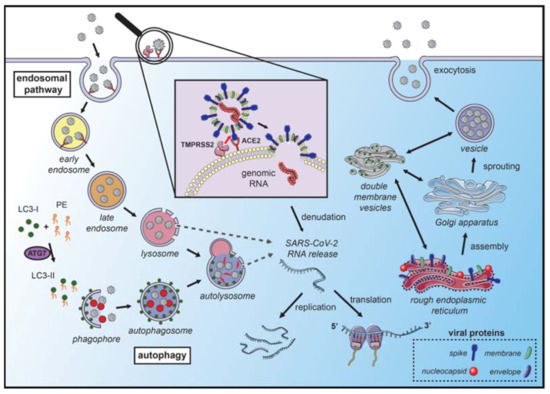
Figure 1. Coronavirus hijacks autophagy machinery to promote their replication. SARS-CoVs bind to the angiotensin-converting enzyme 2 (ACE2) receptor on the membrane surface and enter the host cell. The fusion with the membrane and the release of the genomic RNA into the cytoplasm occurs after the cleavage of the spike (S) protein, which can occur in several locations. S protein cleavage occurs on the cell membrane surface by the transmembrane protease serine 2 (TMPRSS2), which is associated with the ACE2 receptor, or by cathepsin-L and cysteine proteases in the endosomal system. The acidic pH in the lysosomes is necessary for the activity of cathepsin-L and S protein cleavage. Next, the endosomal cargo converges with the autophagic vacuoles in the lysosomes. Coronavirus nonstructural proteins colocalize with microtubule-associated proteins 1A/1B light chain 3A (LC3-II) in the endomembrane system, suggesting that autophagy plays a role in amplifying coronavirus replication. After fusion with the membrane, the genomic RNA is released and stripped of the nucleocapsid protein. Viral proteins are translated in the endoplasmic reticulum, which promotes the rearrangement of endoplasmic reticulum membranes and the formation of double-membrane vesicles, which are also localized with LC3 and autophagy-related proteins. The newly synthesized genomic RNA is then assembled into virions in intermediate compartments located between the endoplasmic reticulum and the Golgi apparatus and moves through the secretory pathway of the host and eventually released by exocytosis (the illustration was produced using the smart servier medical art vectors for publications and presentations licensed under the Creative Commons (CC BY 3.0)) [19].
Table 1. Molecular machinery recruited in autophagy initiation.
| Acronym | Protein | Function | Ref. |
|---|---|---|---|
| 1. Transcriptional factors | |||
| TFEB | Transcription factor EB | A master gene regulator of lysosomal biogenesis and autophagy | [7][8] |
| 2. Initiation of autophagy | |||
| mTORC1 | Mammalian target of rapamycin complex 1 | Nutrient sensor and controller of protein synthesis and autophagy | [20] |
| 3. Upstream regulators of mTORC1 | |||
| AKT | Serine-threonine kinase | Cell growth, proliferation, differentiation and survival signalling | [21][22] |
| AMPK | Adenosine monophosphate-activated protein kinase | Energy homeostasis signalling | [23] |
| BCL-2 | B-cell lymphoma 2 | Regulation of cell death | [4][5] |
| ERK/MAPK | Extracellular signal-regulated kinase/mitogen-activated protein kinase | Regulation of cell proliferation | [14] |
| PI3K | Phosphoinositide 3-kinase | Cell growth, proliferation, differentiation and survival signalling | [21][22] |
| 4. Nucleation and phagophore formation | |||
| Ambra1 | Activating molecule in Beclin-1-regulated autophagy | Positive regulator of Beclin-1-mediated autophagy | [6] |
| BECN1 | Beclin-1 | Regulator of autophagic programmed cell death | [4][5] |
| ULK1 | Unc-51 like autophagy activating kinase | Autophagy initiator | [24][25] |
| 5. Autophagosome formation and elongation | |||
| Atg | Autophagy-related protein | Factors required for the formation of autophagosomal membranes | [20] |
| LC3 | Microtubule-associated proteins 1A/1B light chain 3A | Autophagosomal marker that mediates the physical interactions between microtubules and components of the cytoskeleton | [26] |
| p62/SQSTM1 | Ubiquitin-binding protein p62/Sequestosome-1 | An autophagosome cargo protein that targets and labels other proteins for selective autophagy | [9][27] |
| Vps34 | Vacuolar protein sorting 34 | A class III phosphoinositide 3-kinase that acts on vesicle trafficking | [26] |
| WIPI2 | WD repeat domain phosphoinositide-interacting protein proteins | Regulates the assembly of multiprotein complexes | [26] |
| 6. Autophagosome-lysosome fusion | |||
| SNAP29 | Synaptosome-associated protein 29 | Mediates autophagosome-lysosome fusion | [28] |
| SNARE | N-ethylmaleimide-sensitive factor attachment protein receptor complexes | Vesicle fusion mediator | [28][27] |
| Stx17 | Syntaxin 17 | A SNARE like protein that mediates autophagosome-lysosome fusion | [28][27] |
| VAMP8 | Vesicle-associated membrane protein 8 | A SNARE like protein that mediates autophagosome-lysosome fusion | [28] |
3. Autophagy-Related Therapeutic Targets for COVID-19 Management
3.1. Lysosomotropic Agents
Chloroquine (CQ) and hydroxychloroquine (HCQ) are weak diprotic bases used as antimalarial drugs (Figure 2). These compounds accumulate in the endosome–lysosomal network of cells and neutralize the acidic pH, with consequent blockage of cathepsin activity and lysosomal fusion [29][30]. Previous studies showed that CQ displays a wide-spectrum of antiviral effects against CoVs, chronic HIV, and influenza viruses type A and B, both in vitro and in vivo [31][32].
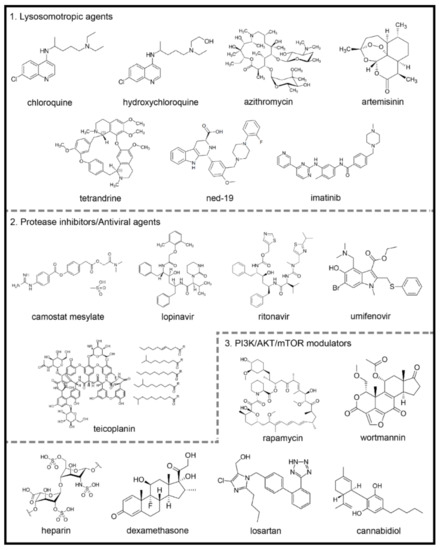
Figure 2. Chemical structures of potential autophagy-related drugs for SARS-CoV-2 infection. The drugs were divided in three groups according to their effects on the autophagy signaling pathway and possible effect against SARS-CoV-2 infection. The lysosomotropic agents (1) can prevent coronavirus infection by alkalinizing the acid pH in the endolysosomal system; some examples are chloroquine, hydroxychloroquine, azithromycin, artemisinin, two-pore channel antagonists (such as tetrandrine and ned-19) and imatinib. The protease inhibitors/antiviral agents (2) can inhibit the proteolytic cleavage of the spike coronavirus protein, which is necessary for viral entry into host cells; some examples are camostat mesylate, lopinavir, ritonavir, umifenovir and teicoplanin. The third group is composed by PI3K/AKT/mTOR signaling pathways modulators (3), which can modulate intracellular pathways related to autophagy and coronavirus infection; some examples are the rapamycin, wortmannin, the anticoagulant heparin, the glucocorticoid dexamethasone, losartan and cannabidiol. The figures for each chemical structure are from according to Wikimedia Commons (Public Domain).
Azithromycin is a broad-spectrum macrolide antibiotic that binds to the S50 ribosomal subunit of bacteria inhibiting its protein synthesis [33] (Figure 2). The antiviral efficacy of azithromycin has been demonstrated in different viral infections [34][35][36][37].
Artemisinin is isolated from the herb Artemisia annua L. [38]. The derivative compounds are sesquiterpene lactones with a unique endoperoxide bridge moiety primarily responsible for their biological actions [39] (Figure 2). ArtemiC, a micellar formulation of artemisinin, curcumin, frankincense (Boswellia) and vitamin C, which is administered by spraying, is in phase II of clinical trials for patients diagnosed with COVID-19 (NIH-Clinical Trials Database; Identifier: NCT04382040 and NCT04553705) [40].
Nicotinic acid adenine dinucleotide phosphate (NAADP) is an intracellular messenger that plays a vital role in the mobilization of Ca2+ in mammalians cells [41][42][43][44] by binding to two-pore channels (TPCs) [45][46]. Furthermore, NAADP has been reported as a potent Ca2+ mobilizing messenger and inducer of autophagy [47][48][49][50][51][52]. On the other hand, the TPC antagonists ned-19 and tetrandine (Figure 2) were postulated as possible blockers of lysosomal function, causing a further inhibition of autophagy on the degradation step [48].
Imatinib, a tyrosine kinase inhibitor developed in 2001, revolutionized the treatment of chronic myeloid leukemia [53], since its activity against the breakpoint cluster region gene-Abelson proto-oncogene (BCR-ABL) in cancerous cells [54] (Figure 2).
3.2. Protease Inhibitors/Antiviral Agents: The Prevention of Infection
3.2.1. Camostat Mesylate
Camostat mesylate inhibits the serine protease TMPRSS2 and prevents the entry of SARS-CoV-2 into the host cells [55] (Figure 2). Other proteases, including cathepsin-L, thermolysins, plasmins and trypsin, can act as a cofactor for virus entry into the host cell [56].
Lopinavir (ABT-378) is a potent protease inhibitor used to prevent HIV replication and spread [57] (Figure 2). It has been suggested that since SARS-CoV-2 contains structural components that are similar to other viruses, including HIV, it is plausible that this antiviral therapy could be used to treat patients with COVID-19 [58].
Umifenovir is currently used in Russia and China as a prophylaxis for the treatment of pulmonary infections caused by human influenza A and B viruses and HCV [59][60] (Figure 2). The proposed mode of action of umifenovir involves intercalation with membrane lipids, inhibiting viral fusion with the plasma membrane of the host cell. It has also been shown that the drug can bind to the membrane-bound clathrin protein and prevent endocytosis of the virus [59].
Teicoplanin is a clinically approved glycopeptide antibiotic that inhibits cathepsin L activity and blocks MERS-CoV and SARS-CoV entry into cells [61] (Figure 2).
3.2.2. PI3K/AKT/mTOR Modulators
Rapamycin is a PI3K/AKT/mTOR inhibitor and clinically proven macrolide that exhibits potent antitumor and immunosuppressive activity [62][63] (Figure 2). While the antiviral activity of rapamycin is controversial [62], it was capable of reducing porcine epidemic diarrhea virus [64], transmissible gastroenteritis virus (TGEV) and CoVs infectivity [65].
Heparin exhibited several antiviral actions [66][67][68][69], probably due to its structural similarity to heparan sulfate [70], a glycosaminoglycan formed by proteoglycans present on the surface of cells that participates in viral entry into eukaryotic cells as an initial anchoring domain [66][71] (Figure 2). Thus, heparan sulfate appears to modulate the entry of SARS-CoV into cells.
Glucocorticoids (GCs) are steroid hormones with potent anti-inflammatory and immunosuppressive actions used in the treatment of chronic inflammatory, autoimmune and allergic diseases [72][73] (Figure 2). During the SARS-CoV and MERS-CoV epidemics, GCs were widely used to decrease the exacerbated immune response caused by the uncontrolled release of proinflammatory cytokines observed during severe lung inflammation [74][75].
Several studies have shown that renin-angiotensin system (RAS) deregulation may be responsible for acute respiratory distress syndrome, which can be triggered by viruses (SARS-CoV, H5N1 and H7N9), bacteria and particles and molecules [76]. Therefore, excess angiotensin II may be primarily responsible for increased SARS-CoV pathogenesis [77]. Thus, these studies suggest that decreasing the angiotensin II levels or blocking the RAS pathway might attenuate acute lung injury severity.
It is known that SARS-CoV-2 infection leads to a proinflammatory cytokine storm [78]; thus, cannabidiol might decrease the levels of these cytokines and benefit patients infected with SARS-CoV-2 [79].
References
- Neilson, S. Aylin Woodward Coronavirus: A 1-Year Timeline of the Pandemic Since China’s 1st Case—Business Insider. Available online: (accessed on 24 December 2020).
- Sarkar, R.; Mitra, S.; Chandra, P.; Saha, P.; Banerjee, A.; Dutta, S.; Chawla-Sarkar, M. Comprehensive analysis of genomic diversity of SARS-CoV-2 in different geographic regions of India: An endeavour to classify Indian SARS-CoV-2 strains on the basis of co-existing mutations. Arch. Virol. 2021.
- Gómez-Carballa, A.; Bello, X.; Pardo-Seco, J.; Martinón-Torres, F.; Salas, A. Mapping genome variation of SARS-CoV-2 worldwide highlights the impact of COVID-19 super-spreaders. Genome Res. 2020, 30, 1434–1448.
- He, C.; Klionsky, D.J. Regulation Mechanisms and Signaling Pathways of Autophagy. Annu. Rev. Genet. 2009, 43, 67–93.
- He, C.; Levine, B. The Beclin 1 interactome. Curr. Opin. Cell Biol. 2010, 22, 140–149.
- Fimia, G.M.; Corazzari, M.; Antonioli, M.; Piacentini, M. Ambra1 at the crossroad between autophagy and cell death. Oncogene 2013, 32, 3311–3318.
- Yue, Y.; Nabar, N.R.; Shi, C.S.; Kamenyeva, O.; Xiao, X.; Hwang, I.Y.; Wang, M.; Kehrl, J.H. SARS-Coronavirus Open Reading Frame-3a drives multimodal necrotic cell death. Cell Death Dis. 2018, 9, 1–15.
- Shi, B.; Ma, M.; Zheng, Y.; Pan, Y.; Lin, X. mTOR and Beclin1: Two key autophagy-related molecules and their roles in myocardial ischemia/reperfusion injury. J. Cell. Physiol. 2019, 234, 12562–12568.
- Orvedahl, A.; MacPherson, S.; Sumpter, R.; Tallóczy, Z.; Zou, Z.; Levine, B. Autophagy Protects against Sindbis Virus Infection of the Central Nervous System. Cell Host Microbe 2010, 7, 115–127.
- Ojha, C.R.; Rodriguez, M.; Karuppan, M.K.M.; Lapierre, J.; Kashanchi, F.; El-Hage, N. Toll-like receptor 3 regulates Zika virus infection and associated host inflammatory response in primary human astrocytes. PLoS ONE 2019, 14, e0208543.
- Karuppan, M.K.M.; Ojha, C.R.; Rodriguez, M.; Lapierre, J.; Aman, M.J.; Kashanchi, F.; Toborek, M.; Nair, M.; El-Hage, N. Reduced-Beclin1-Expressing Mice Infected with Zika-R103451 and Viral-Associated Pathology during Pregnancy. Viruses 2020, 12, 608.
- Chen, X.; Wang, K.; Xing, Y.; Tu, J.; Yang, X.; Zhao, Q.; Li, K.; Chen, Z. Coronavirus membrane-associated papain-like proteases induce autophagy through interacting with Beclin1 to negatively regulate antiviral innate immunity. Protein Cell 2014.
- Gassen, N.C.; Niemeyer, D.; Muth, D.; Corman, V.M.; Martinelli, S.; Gassen, A.; Hafner, K.; Papies, J.; Mösbauer, K.; Zellner, A.; et al. SKP2 attenuates autophagy through Beclin1-ubiquitination and its inhibition reduces MERS-Coronavirus infection. Nat. Commun. 2019.
- Kindrachuk, J.; Ork, B.; Hart, B.J.; Mazur, S.; Holbrook, M.R.; Frieman, M.B.; Traynor, D.; Johnson, R.F.; Dyall, J.; Kuhn, J.H.; et al. Antiviral potential of ERK/MAPK and PI3K/AKT/mTOR signaling modulation for Middle East respiratory syndrome coronavirus infection as identified by temporal kinome analysis. Antimicrob. Agents Chemother. 2015, 59, 1088–1099.
- Gassen, N.C.; Papies, J.; Bajaj, T.; Dethloff, F.; Emanuel, J.; Weckmann, K.; Heinz, D.E.; Heinemann, N.; Lennarz, M.; Richter, A.; et al. Analysis of SARS-CoV-2-controlled autophagy reveals spermidine, MK-2206, and niclosamide as putative antiviral therapeutics. bioRxiv 2020.
- Yang, N.; Shen, H.M. Targeting the endocytic pathway and autophagy process as a novel therapeutic strategy in COVID-19. Int. J. Biol. Sci. 2020, 16, 1724–1731.
- Benvenuto, D.; Angeletti, S.; Giovanetti, M.; Bianchi, M.; Pascarella, S.; Cauda, R.; Ciccozzi, M.; Cassone, A. Evolutionary analysis of SARS-CoV-2: How mutation of Non-Structural Protein 6 (NSP6) could affect viral autophagy. J. Infect. 2020, 81, e24–e27.
- Mercatelli, D.; Holding, A.N.; Giorgi, F.M. Web tools to fight pandemics: The COVID-19 experience. Brief. Bioinform. 2020, 2020, 1–11.
- Servier Medical Art by Servier Is Licensed under a Creative Commons Attribution 3.0 Unported License. Servier Medical Art. Available online: (accessed on 2 January 2021).
- Kim, Y.C.; Guan, K.L. MTOR: A pharmacologic target for autophagy regulation. J. Clin. Investig. 2015, 125, 25–32.
- Inoki, K.; Li, Y.; Zhu, T.; Wu, J.; Guan, K.L. TSC2 is phosphorylated and inhibited by Akt and suppresses mTOR signalling. Nat. Cell Biol. 2002, 4, 648–657.
- Laplante, M.; Sabatini, D.M. Regulation of mTORC1 and its impact on gene expression at a glance. J. Cell Sci. 2013, 126, 1713–1719.
- Gwinn, D.M.; Shackelford, D.B.; Egan, D.F.; Mihaylova, M.M.; Mery, A.; Vasquez, D.S.; Turk, B.E.; Shaw, R.J. AMPK Phosphorylation of Raptor Mediates a Metabolic Checkpoint. Mol. Cell 2008, 30, 214–226.
- Ganley, I.G.; Lam, D.H.; Wang, J.; Ding, X.; Chen, S.; Jiang, X. ULK1·ATG13·FIP200 complex mediates mTOR signaling and is essential for autophagy. J. Biol. Chem. 2009, 284, 12297–12305.
- Jung, C.H.; Jun, C.B.; Ro, S.H.; Kim, Y.M.; Otto, N.M.; Cao, J.; Kundu, M.; Kim, D.H. ULK-Atg13-FIP200 complexes mediate mTOR signaling to the autophagy machinery. Mol. Biol. Cell 2009, 20, 1992–2003.
- Polson, H.E.J.; De Lartigue, J.; Rigden, D.J.; Reedijk, M.; Urbé, S.; Clague, M.J.; Tooze, S.A. Mammalian Atg18 (WIPI2) localizes to omegasome-anchored phagophores and positively regulates LC3 lipidation. Autophagy 2010, 6, 506–522.
- Tian, L.; Yang, Y.; Li, C.; Chen, J.; Li, Z.; Li, X.; Li, S.; Wu, F.; Hu, Z.; Yang, Z. The cytotoxicity of coxsackievirus B3 is associated with a blockage of autophagic flux mediated by reduced syntaxin 17 expression article. Cell Death Dis. 2018, 9, 1–12.
- Uematsu, M.; Nishimura, T.; Sakamaki, Y.; Yamamoto, H.; Mizushima, N. Accumulation of undegraded autophagosomes by expression of dominant-negative STX17 (syntaxin 17) mutants. Autophagy 2017, 13, 1452–1464.
- Al-Bari, M.A.A. Targeting endosomal acidification by chloroquine analogs as a promising strategy for the treatment of emerging viral diseases. Pharmacol. Res. Perspect. 2017, 5.
- Mauthe, M.; Orhon, I.; Rocchi, C.; Zhou, X.; Luhr, M.; Hijlkema, K.J.; Coppes, R.P.; Engedal, N.; Mari, M.; Reggiori, F. Chloroquine inhibits autophagic flux by decreasing autophagosome-lysosome fusion. Autophagy 2018, 14, 1435–1455.
- Vincent, M.J.; Bergeron, E.; Benjannet, S.; Erickson, B.R.; Rollin, P.E.; Ksiazek, T.G.; Seidah, N.G.; Nichol, S.T. Chloroquine is a potent inhibitor of SARS coronavirus infection and spread. Virol. J. 2005, 2, 69.
- Savarino, A.; Di Trani, L.; Donatelli, I.; Cauda, R.; Cassone, A. New insights into the antiviral effects of chloroquine. Lancet Infect. Dis. 2006, 6, 67–69.
- Parnham, M.J.; Haber, V.E.; Giamarellos-Bourboulis, E.J.; Perletti, G.; Verleden, G.M.; Vos, R. Azithromycin: Mechanisms of action and their relevance for clinical applications. Pharmacol. Ther. 2014, 143, 225–245.
- Gielen, V.; Johnston, S.L.; Edwards, M.R. Azithromycin induces anti-viral responses in bronchial epithelial cells. Eur. Respir. J. 2010, 36, 646–654.
- Madrid, P.B.; Panchal, R.G.; Warren, T.K.; Shurtleff, A.C.; Endsley, A.N.; Green, C.E.; Kolokoltsov, A.; Davey, R.; Manger, I.D.; Gilfillan, L.; et al. Evaluation of Ebola Virus Inhibitors for Drug Repurposing. ACS Infect. Dis. 2016, 1, 317–326.
- Retallack, H.; Di Lullo, E.; Arias, C.; Knopp, K.A.; Laurie, M.T.; Sandoval-Espinosa, C.; Leon, W.R.M.; Krencik, R.; Ullian, E.M.; Spatazza, J.; et al. Zika virus cell tropism in the developing human brain and inhibition by azithromycin. Proc. Natl. Acad. Sci. USA 2016, 113, 14408–14413.
- Tran, D.H.; Sugamata, R.; Hirose, T.; Suzuki, S.; Noguchi, Y.; Sugawara, A.; Ito, F.; Yamamoto, T.; Kawachi, S.; Akagawa, K.S.; et al. Azithromycin, a 15-membered macrolide antibiotic, inhibits influenza A(H1N1)pdm09 virus infection by interfering with virus internalization process. J. Antibiot. 2019, 72, 759–768.
- Medhi, B.; Patyar, S.; Rao, R.S.; Byrav DS, P.; Prakash, A. Pharmacokinetic and Toxicological Profile of Artemisinin Compounds: An Update. Pharmacology 2009, 84, 323–332.
- Sun, X.; Yan, P.; Zou, C.; Wong, Y.; Shu, Y.; Lee, Y.M.; Zhang, C.; Yang, N.; Wang, J.; Zhang, J. Targeting autophagy enhances the anticancer effect of artemisinin and its derivatives. Med. Res. Rev. 2019, 39, 2172–2193.
- U.S. Department of Health & Human Services. Available online: (accessed on 2 January 2021).
- Lee, H.C.; Aarhus, R. Functional visualization of the separate but interacting calcium stores sensitive to NAADP and cyclic ADP-ribose. J. Cell Sci. 2000, 113, 4413–4420.
- Patel, S.; Churchill, G.C.; Galione, A. Coordination of Ca2+ signalling by NAADP. Trends Biochem. Sci. 2001, 26, 482–489.
- Galione, A. NAADP, a new intracellular messenger that mobilizes Ca2+ from acidic stores. Biochem. Soc. Trans. 2006, 34, 922–926.
- Ruas, M.; Rietdorf, K.; Arredouani, A.; Davis, L.C.; Lloyd-Evans, E.; Koegel, H.; Funnell, T.M.; Morgan, A.J.; Ward, J.A.; Watanabe, K.; et al. Purified TPC Isoforms Form NAADP Receptors with Distinct Roles for Ca2+ Signaling and Endolysosomal Trafficking. Curr. Biol. 2010, 20, 703–709.
- Brailoiu, E.; Churamani, D.; Cai, X.; Schrlau, M.G.; Brailoiu, G.C.; Gao, X.; Hooper, R.; Boulware, M.J.; Dun, N.J.; Marchant, J.S.; et al. Essential requirement for two-pore channel 1 in NAADP-mediated calcium signaling. J. Cell Biol. 2009, 186, 201–209.
- Calcraft, P.J.; Ruas, M.; Pan, Z.; Cheng, X.; Arredouani, A.; Hao, X.; Tang, J.; Rietdorf, K.; Teboul, L.; Chuang, K.-T.; et al. NAADP mobilizes calcium from acidic organelles through two-pore channels. Nature 2009, 459, 596–600.
- Pereira, G.J.S.; Hirata, H.; Fimia, G.M.; Do Carmo, L.G.; Bincoletto, C.; Han, S.W.; Stilhano, R.S.; Ureshino, R.P.; Bloor-Young, D.; Churchill, G.; et al. Nicotinic Acid Adenine Dinucleotide Phosphate (NAADP) regulates autophagy in cultured astrocytes. J. Biol. Chem. 2011, 286, 27875–27881.
- Pereira, G.J.S.; Antonioli, M.; Hirata, H.; Ureshino, R.P.; Nascimento, A.R.; Bincoletto, C.; Vescovo, T.; Piacentini, M. Glutamate induces autophagy via the two-pore channels in neural cells. Oncotarget 2017, 8, 12730–12740.
- Pereira, G.J.S.; Hirata, H.; do Carmo, L.G.; Stilhano, R.S.; Ureshino, R.P.; Medaglia, N.C.; Han, S.W.; Churchill, G.; Bincoletto, C.; Patel, S.; et al. NAADP-sensitive two-pore channels are present and functional in gastric smooth muscle cells. Cell Calcium 2014, 56, 51–58.
- Gómez-Suaga, P.; Luzón-Toro, B.; Churamani, D.; Zhang, L.; Bloor-Young, D.; Patel, S.; Woodman, P.G.; Churchill, G.C.; Hilfiker, S. Leucine-rich repeat kinase 2 regulates autophagy through a calcium-dependent pathway involving NAADP. Hum. Mol. Genet. 2012, 21, 511–525.
- Rah, S.; Lee, Y.; Kim, U. NAADP-mediated Ca2+ signaling promotes autophagy and protects against LPS-induced liver injury. FASEB J. 2017, 31, 3126–3137.
- Ogunbayo, O.A.; Duan, J.; Xiong, J.; Wang, Q.; Feng, X.; Ma, J.; Zhu, M.X.; Evans, A.M. MTORC1 controls lysosomal Ca2+ release through the two-pore channel TPC2. Sci. Signal. 2018, 11, 5775.
- Iqbal, N.; Iqbal, N. Imatinib: A Breakthrough of Targeted Therapy in Cancer. Chemother. Res. Pract. 2014, 2014, 357027.
- Sisk, J.M.; Frieman, M.B.; Machamer, C.E. Coronavirus S protein-induced fusion is blocked prior to hemifusion by Abl kinase inhibitors. J. Gen. Virol. 2018, 99, 619–630.
- Hoffmann, M.; Kleine-Weber, H.; Schroeder, S.; Krüger, N.; Herrler, T.; Erichsen, S.; Schiergens, T.S.; Herrler, G.; Wu, N.H.; Nitsche, A.; et al. SARS-CoV-2 Cell Entry Depends on ACE2 and TMPRSS2 and Is Blocked by a Clinically Proven Protease Inhibitor. Cell 2020, 181, 271–280.e8.
- Rahman, N.; Basharat, Z.; Yousuf, M.; Castaldo, G.; Rastrelli, L.; Khan, H. Virtual Screening of Natural Products against Type II Transmembrane Serine Protease (TMPRSS2), the Priming Agent of Coronavirus 2 (SARS-CoV-2). Molecules 2020, 25, 2271.
- McKee, D.L.; Sternberg, A.; Stange, U.; Laufer, S.; Naujokat, C. Candidate drugs against SARS-CoV-2 and COVID-19. Pharmacol. Res. 2020, 157, 104859.
- Barlow, A.; Landolf, K.M.; Barlow, B.; Yeung, S.Y.A.; Heavner, J.J.; Claassen, C.W.; Heavner, M.S. Review of Emerging Pharmacotherapy for the Treatment of Coronavirus Disease 2019. Pharmacotherapy 2020, 40, 416–437.
- Boriskin, Y.S.; Leneva, I.A.; Pécheur, E.-I.; Polyak, S.J. Arbidol: A Broad-Spectrum Antiviral Compound that Blocks Viral Fusion. Curr. Med. Chem. 2008, 15, 997–1005.
- Boriskin, Y.S.; Pécheur, E.I.; Polyak, S.J. Arbidol: A broad-spectrum antiviral that inhibits acute and chronic HCV infection. Virol. J. 2006, 3.
- Zhou, N.; Pan, T.; Zhang, J.; Li, Q.; Zhang, X.; Bai, C.; Huang, F.; Peng, T.; Zhang, J.; Liu, C.; et al. Glycopeptide antibiotics potently inhibit cathepsin l in the late endosome/lysosome and block the entry of ebola virus, middle east respiratory syndrome coronavirus (MERS-CoV), and severe acute respiratory syndrome coronavirus (SARS-CoV). J. Biol. Chem. 2016, 291, 9218–9232.
- Maiese, K. The mechanistic target of rapamycin (mTOR): Novel Considerations as an Antiviral Treatment and Possibilities for COVID-19. Curr. Neurovasc. Res. 2020, 17.
- Pandrea, I.; Landay, A.L. Implications for Therapy. In Models of Protection Against HIV/SIV; Elsevier Inc.: Amsterdam, The Netherlands, 2012; pp. 81–132. ISBN 9780123877154.
- Ko, S.; Gu, M.J.; Kim, C.G.; Kye, Y.C.; Lim, Y.; Lee, J.E.; Park, B.C.; Chu, H.; Han, S.H.; Yun, C.H. Rapamycin-induced autophagy restricts porcine epidemic diarrhea virus infectivity in porcine intestinal epithelial cells. Antivir. Res. 2017, 146, 86–95.
- Guo, L.; Yu, H.; Gu, W.; Luo, X.; Li, R.; Zhang, J.; Xu, Y.; Yang, L.; Shen, N.; Feng, L.; et al. Autophagy Negatively Regulates Transmissible Gastroenteritis Virus Replication. Sci. Rep. 2016, 6, 1–14.
- Shukla, D.; Spear, P.G. Herpesviruses and heparan sulfate: An intimate relationship in aid of viral entry. J. Clin. Investig. 2001, 108, 503–510.
- Basu, A.; Kanda, T.; Beyene, A.; Saito, K.; Meyer, K.; Ray, R. Sulfated Homologues of Heparin Inhibit Hepatitis C Virus Entry into Mammalian Cells. J. Virol. 2007, 81, 3933–3941.
- Ghezzi, S.; Cooper, L.; Rubio, A.; Pagani, I.; Rosaria, M.; Ippolito, G.; Pelletier, J.; Yates, E.A.; Vicenzi, E. Heparin prevents Zika virus induced-cytopathic effects in human neural progenitor cells Silvia. Antivir. Res. 2020, 140, 13–17.
- Vicenzi, E.; Canducci, F.; Pinna, D.; Mancini, N.; Carletti, S.; Lazzarin, A.; Bordignon, C.; Poli, G.; Clementi, M. Coronaviridae and SARS-associated Coronavirus Strain HSR1. Emerg. Infect. Dis. 2004, 10, 413–418.
- Sardu, C.; Gambardella, J.; Morelli, M.B.; Wang, X.; Marfella, R.; Santulli, G. Hypertension, Thrombosis, Kidney Failure, and Diabetes: Is COVID-19 an Endothelial Disease? A Comprehensive Evaluation of Clinical and Basic Evidence. J. Clin. Med. 2020, 9, 1417.
- Lang, J.; Yang, N.; Deng, J.; Liu, K.; Yang, P.; Zhang, G.; Jiang, C. Inhibition of SARS pseudovirus cell entry by lactoferrin binding to heparan sulfate proteoglycans. PLoS ONE 2011, 6, e23710.
- Vandewalle, J.; Luypaert, A.; De Bosscher, K.; Libert, C. Therapeutic Mechanisms of Glucocorticoids. Trends Endocrinol. Metab. 2018, 29, 42–54.
- Sundahl, N.; Bridelance, J.; Libert, C.; De Bosscher, K.; Beck, I.M. Selective glucocorticoid receptor modulation: New directions with non-steroidal scaffolds. Pharmacol. Ther. 2015, 152, 28–41.
- Russell, C.D.; Millar, J.E.; Baillie, J.K. Clinical evidence does not support corticosteroid treatment for 2019-nCoV lung injury. Lancet 2020, 395, 473–475.
- Zhang, W.; Zhao, Y.; Zhang, F.; Wang, Q.; Li, T.; Liu, Z.; Wang, J.; Qin, Y.; Zhang, X.; Yan, X.; et al. The use of anti-inflammatory drugs in the treatment of people with severe coronavirus disease 2019 (COVID-19): The Perspectives of clinical immunologists from China. Clin. Immunol. 2020, 214, 108393.
- Zou, Z.; Yan, Y.; Shu, Y.; Gao, R.; Sun, Y.; Li, X.; Ju, X.; Liang, Z.; Liu, Q.; Zhao, Y.; et al. Angiotensin-converting enzyme 2 protects from lethal avian influenza A H5N1 infections. Nat. Commun. 2014, 5, 1–7.
- Huang, F.; Guo, J.; Zou, Z.; Liu, J.; Cao, B.; Zhang, S.; Li, H.; Wang, W.; Sheng, M.; Liu, S.; et al. Angiotensin II plasma levels are linked to disease severity and predict fatal outcomes in H7N9-infected patients. Nat. Commun. 2014, 5, 3595.
- Giamarellos-Bourboulis, E.J.; Netea, M.G.; Rovina, N.; Akinosoglou, K.; Antoniadou, A.; Antonakos, N.; Damoraki, G.; Gkavogianni, T.; Adami, M.E.; Katsaounou, P.; et al. Complex Immune Dysregulation in COVID-19 Patients with Severe Respiratory Failure. Cell Host Microbe 2020, 27, 992–1000.e3.
- Byrareddy, S.N.; Mohan, M. SARS-CoV2 induced respiratory distress: Can cannabinoids be added to anti-viral therapies to reduce lung inflammation? Brain Behav. Immun. 2020, 87, 120.

