 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Raffaella Rametta | + 7682 word(s) | 7682 | 2020-05-20 08:55:07 | | | |
| 2 | Camila Xu | Meta information modification | 7682 | 2020-05-26 10:43:07 | | | | |
| 3 | Rita Xu | -68 word(s) | 7614 | 2020-10-28 10:03:15 | | |
Video Upload Options
The environment and the human genome are closely entangled and many genetic variations that occur in human populations are the result of adaptive selection to ancestral environmental (mainly dietary) conditions. However, the selected mutations may become maladaptive when environmental conditions change, thus becoming candidates for diseases. Hereditary hemochromatosis (HH) is a potentially lethal disease leading to iron accumulation mostly due to mutations in the HFE gene. Indeed, homozygosity for the C282Y HFE mutation is associated with the primary iron overload phenotype. However, both penetrance of the C282Y variant and the clinical manifestation of the disease are extremely variable, suggesting that other genetic, epigenetic and environmental factors play a role in the development of HH, as well as, and in its progression to end-stage liver diseases. Alcohol consumption and dietary habits may impact on the phenotypic expression of HFE-related hemochromatosis. Indeed, dietary components and bioactive molecules can affect iron status both directly by modulating its absorption during digestion and indirectly by the epigenetic modification of genes involved in its uptake, storage and recycling. Thus, the premise of this review is to discuss how environmental pressures led to the selection of HFE mutations and whether nutritional and lifestyle interventions may exert beneficial effects on HH outcomes and comorbidities.
1. When Diet and Lifestyle May Change Our Genes
Hereditary hemochromatosis (HH) represents one of the most important examples of diseases induced by maladaptive mutations that have been selected during evolutionary processes. It is a common autosomal recessive disorder characterized by excessive iron deposition in the liver and extrahepatic tissues due to inappropriately high iron absorption. Early symptoms of HH may include weakness, lethargy, arthralgias and impotence. Later manifestations include cirrhosis, hepatocellular cancer (HCC), osteoporosis, cardiomyopathy, dysrhythmia, diabetes (T2D) and hypogonadism. Currently, phlebotomy represents the main therapeutic option for HH treatment [1].
Hepcidin (HAMP) is the hepatic hormone that regulates iron absorption. It controls systemic iron availability though the negative regulation of its target ferroportin (FPN), a transmembrane iron efflux channel highly expressed in enterocytes, macrophages and hepatocytes. Hepcidin regulators are activated by positive stimuli (iron status and inflammation), and negative ones (erythropoietic activity and hypoxia). Mutations occurring in genes involved in hepcidin expression and regulation, which cause a defective production or activity of the hormone, lead to HH [2] [3].
Most cases of HH are attributable to mutations in the HFE gene, which encodes a non-classical MHC class 1 protein involved in the downregulation of iron absorption [4]. HFE, which is predominantly located at the hepatocyte cell surface, competes with transferrin (Tf) to bind transferrin receptor-1 (TfR1) thus reducing TfR1–Tf interactions and negatively regulating iron uptake. With increasing plasma iron levels, the iron-loaded Tf (holo-Tf) gains high affinity for TfR1, HFE is displaced from the HFE-TfR1 complex and becomes available to associate with transferrin receptor-2 (TfR2) and hemojuvelin (HJV). The formed HFE–TfR2–HJV complex triggers the bone morphogenetic protein (BMP)/SMAD signaling pathway to hepcidin gene (HAMP) expression [5].
HFE is also expressed in intestinal enterocytes. Indeed, HFE was found as a complex with TfR1 on the basolateral membrane of enterocytes in the duodenal crypt cells, as well as, in the villus enterocytes of the small intestine. It has been speculated that HFE may act as a plasma iron sensor by modulating Tf-mediated uptake or the release of dietary iron by these cells [6].
The transition c.845G > A (rs1800562), resulting in the amino acid substitution from cysteine to tyrosine at position 282 (C282Y) in the HFE protein, blocks the ability of HFE to downregulate iron absorption by preventing its expression on the cell surface. C282Y homozygosity is associated with iron primary overload phenotype in more than 60% of Europeans [7]. The transversion c.187C > G (rs1799945), which leads to the histidine-to-aspartic acid substitution at position 63 (H63D), has a milder effect on iron absorption. Only 5%–7% of HH patients are compound heterozygotes for C282Y/H63D, whereas heterozygosity for C282Y alone or homozygosity for H63D, is rarely encountered in HH [8]. Lastly, the c.193A > T (rs1800730) transversion, which causes the serine to cysteine substitution at position 65 (S65C) in HFE protein, has been involved in the development of a less severe form of hemochromatosis only in combination with C282Y or H63D mutations [9] [10].
The C282Y variant is predominantly enriched in the European population and it is less frequent (Hispanics and Pacific Islanders) or nearly absent (Asians and Africans) in non-Caucasian populations [11]. In addition, the prevalence of C282Y homozygosity follows a strong geographical distribution among Europeans with the highest frequencies in Northern Europe (10.9% in Ireland, 9.7% in Scotland, 8.2% in Wales, 7.8% in Brittany, 7.3% in Norway and 7% in Denmark) and the lowest frequencies in southern Europe (ranging from 1 to 5% in the Mediterranean area)[12].
Evolutionary analyses suggest that the C282Y mutation in the HFE gene may have originated from a mutation in a single Celtic or Viking ancestor around 4000 BC [13] and was spread in central Europe following the migratory flows. Distante and colleagues suggested that the spread of the C282Y variant could be the result of an adaptation to a dietary shift from a hunter-gatherer diet based on wild foods (rich in iron) to a Neolithic diet based on cereal and dairy food (poor in iron) [14].
Indeed, before the Neolithic Age, the subsistence strategies were based on wild flora and fauna (game, fish, shellfish, insects, nuts, roots and vegetables). Red meat and many species of shellfish provided a rich source of highly digestible heme-iron [15]. During the Neolithic Age, the introduction of agriculture (mainly of cereal grains and other seeds) and animal breeding caused a very drastic change in eating habits leading to the primary dependence on cereals and dairy products, thus shifting human subsistence toward a high carbohydrate/low iron diet [16]. The combination of grains and bovine milk product consumption reduced effective iron absorption in Neolithic farmers. Indeed, on one hand, phytates localized on the surface of cereal grains and other seeds can chelate dietary iron and several other minerals, making them physiologically unavailable [17][18]. On the other hand, the calcium-rich bovine milk is iron-free as it lacks in lactoferrin and thus it may impair the absorption of non-heme iron [19] [20].
In addition, the sedentary lifestyle imposed by agriculture resulted in increased fertility and rapid population growth with a consequent inter-birth interval reduction, which makes it difficult for women to restore body iron stores between two pregnancies [21][22].
As a consequence of changes in lifestyle and food habits, Neolithic farmers were exposed to the risk of iron deficiency anemia. Hence a C282Y mutation in HFE, leading to increased iron absorption, provides a clear selective advantage and represents the genome adaptive response to the environmental changes [23].
The interaction between Celtic culture diffusion and Neolithic niche spreading throughout Europe, which resulted in the selection of HFE C282Y mutation during the Neolithic Age is schematically represented in Figure 1.
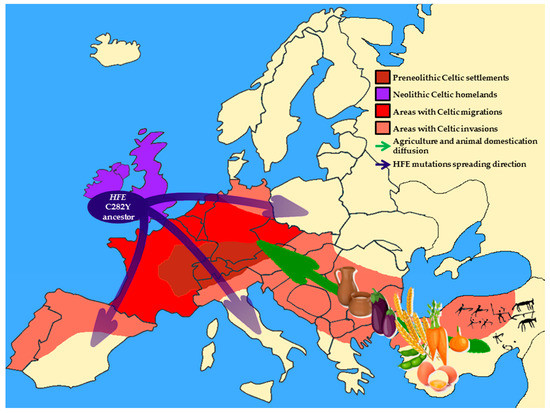
Figure 1. Schematic representation of Europe during the Neolithic Age. Brown area indicates pre-Neolithic original Celtic settlements; purple area shows Celtic homeland during the Neolithic Age; red area is regions occupied by the main Celtic settlements after Neolithic migrations; orange areas indicate the areas reached by the Celtic invasions. Blue arrows show the gradient of HFE C282Y spreading throughout Europe. The green arrow represents the diffusion of agriculture and the domestication of animals.
2. How to Change the Environment to Treat the Disease
Epidemiological studies suggested that the penetrance of the commonest C282Y HFE mutation on iron overload phenotype is extremely variable ranging from 2% to 38% in males and from 1% to 10% in females. Furthermore, there is huge inter-individual variability in the clinical manifestations of HH. Indeed, homozygous individuals may be asymptomatic or they can have a more complicated disease showing hemochromatosis-associated comorbidities (fibrosis or cirrhosis) or other clinically relevant manifestations (T2D, cardiovascular diseases, arthropathy and hypogonadism) [24][25][26]. This evidence supports the notion that other genetic, epigenetic and environmental factors play a role in the development of the disease and in its progression to end-stage liver diseases. Moreover, several environmental and behavioral factors such as diet composition and alcohol consumption may impact on phenotypic expression of HFE-related hemochromatosis.
2.1. Alcohol Consumption
Alcohol abuse is one of the major non-genetic modifiers of iron accumulation in HH patients and may contribute to the progression of symptoms [27]. Indeed, it has been described that excessive alcohol intake precipitates cirrhosis onset in patients with HFE-related hemochromatosis [28]. Moreover, studies conducted in 224 C282Y homozygous patients showed that heavy drinkers (with an alcoholic intake > 60 g dayly) had higher iron indices [27] [29] and a nine-fold increased risk of developing cirrhosis than those consuming safer alcohol doses [27][30][31]. However, the lack of large perspective epidemiological studies reporting the accurate history of alcohol consumption, does not allow for the assessment of the absolute risk of its intake, regardless of the amount ingested, on the HH phenotype.
Nevertheless, in healthy subjects, alcohol consumption reduced hepcidin expression and increased dietary iron absorption, leading to increased serum iron parameters [32][33][34]. In addition, in vitro and in vivo studies suggest a direct role of alcohol in the downregulation of HAMP expression in hepatoma cells and in livers of both short-term alcohol-exposed mice [32] and chronic alcohol-treated rats fed with iron-enriched diets [35][36].
2.2. Dietary Iron Sources: Heme and Non-Heme Dietary Iron
Iron bioavailability and its dietary intake are determinants of iron status in the general population [37][38]. Dietary iron exists as heme and non-heme bound forms, which are absorbed by intestinal epithelial cells through different mechanisms. While heme-iron is taken up intact and then released from the porphyrin ring by heme oxygenase, non-heme iron requires active transport by divalent metal transporter 1 (DMT1) to cross the enterocyte membrane [39][40]. This difference makes heme-iron more readily available than the non-heme one [41]. Indeed, it has been estimated that heme-iron, derived from foods of animal source, accounts for 10%–15% of total iron intake in meat-eating populations. Conversely, the bioavailability of non-heme iron is lower and is closely related to the balance between inhibitors and enhancers of absorption, which are present in foods and in the body iron storages [41].
Nevertheless, the potential diet-related effects on iron accumulation in HH are poorly investigated. C282Y homozygous patients showed increased absorption of both heme- and non-heme iron . Moreover, in the same patients the percentage of non-heme iron absorption from the un-supplemented meal was inversely related to the body iron stores, suggesting that its regulation is impaired in these patients. On the contrary, heme-iron absorption seems to be unaffected by body iron stores. However, when meals were enriched with iron and modified to improve its availability, an exaggerated absorptive response to non-heme iron independently of iron stores was observed in patients’ heterozygous first relatives. These observations emphasize the impact that the introduction of iron-supplemented foods could have in subjects at risk of developing significant iron overload [42].
In a recent systematic review [42], Moretti and colleagues estimated whether and to what extent dietary iron restriction and modulation of dietary iron bioavailability in both C282Y homozygous and idiopathic patients could represent a therapeutic approach in the prevention and management of HH. These authors highlighted that iron bioavailability is 2- to 10-fold higher in patients with clinically overt disease compared to wild-type [43] and it is influenced by the food matrix [44]. However, scant direct evidence is available to support the hypothesis that dietary modulation alone can reduce iron accumulation in HH subjects. Indeed, the results obtained from small cross-sectional studies [45][46][47][48] remain still controversial because of the limited number of subjects included and the potential confounding effects of chronic subclinical inflammation that impact on iron status. Nevertheless, they suggest that a dietary strategy aimed at reducing iron consumption and bioavailability, paralleled by an adequate intake of other essential nutrients, may provide an auxiliary measure to inhibit iron accumulation and reduce the number of required phlebotomy procedures in HH patients [42].
2.3. Insulin Resistance and Iron Homeostasis
Several lines of evidence indicate that ferritin levels and body iron stores should be considered as hallmarks of metabolic syndrome (MetS) whose key pathogenic feature is represented by insulin resistance (IR) [49][50][51]. Patients affected by nonalcoholic fatty liver disease (NAFLD), which is considered the hepatic manifestation of MetS [52], are frequently characterized by mild hepatic iron accumulation [53]. The pathological condition that encompasses fatty liver, increased ferritin levels and enhanced body iron depots in the presence of IR, is referred to as dysmetabolic iron overload syndrome (DIOS). DIOS is observed in 15%–30% of patients with MetS and it is considered the most common iron overload disorder [54].
Excessive iron storages catalyze reactive oxygen species (ROS) overproduction and in turn, lipid peroxidation [55]. In keeping with this data, it has been demonstrated that high-fat diet (HFD) administration induces hepatic iron deposition, paralleled by ferritin induction and by TfR-1 and iron regulated protein-1 (IRP1) overexpression in rodents [56].
Likewise, patients affected by HH often display reduced insulin sensitivity and release, thus increasing their susceptibility to developing NAFLD [53]. In particular, depending on the magnitude of iron depots, around 53%–80% of HH patients develop T2D [57]. In a cross-sectional population-based study, Tuomainen and colleagues revealed a statistically significant alteration of glucose homeostasis in 1013 middle-aged men with hyperferritinemia, in eastern Finland [58]. Moreover, in a case-control study, the risk of developing T2D in subjects with elevated iron stores was attested at 2.4-fold higher than controls [59]. The correlation between serum ferritin levels and T2D has been further confirmed even in 9486 U.S. adults aged > or = 20 years from the Third National Health and Nutrition Examination Survey (NHANES; 1988–1994) [60] and in 27,548 individuals recruited in the European Prospective Investigation of Cancer (EPIC)-Norfolk Cohort Study [61].
The prevalence of the C282Y HFE mutation is significantly increased in patients with NAFLD compared to controls and the difference is more striking in patients with hyperferritinemia than in those without [53]. Furthermore, it has been demonstrated that iron deposition in hepatocytes and in non-parenchymal cells is associated with more severe liver damage in 587 Italian patients with NAFLD carrying the HFE variations [62]. These patients develop NAFLD even in the presence of less severe metabolic abnormalities, suggesting that iron overload may contribute to NAFLD onset and progression towards end-stage liver diseases [62].
Insulin signaling and iron homeostasis are closely entangled, as revealed by a broad number of clinical and preclinical studies [63][64]. Insulin is able to stimulate ferritin synthesis and facilitates iron uptake. In turn, increased iron concentrations lead to peripheral hyperinsulinemia [65]. The likely mechanism through which the iron excess impairs hepatocyte insulin sensitivity is due to its direct interfering with the insulin receptor and intracellular insulin signaling cascade [66]. Moreover, pancreatic β-cells are extremely sensitive to iron-related ROS, thus precipitating T2D and its comorbidities onset [55].
According to this notion, iron depletion through phlebotomy along with lifestyle modifications ameliorate insulin sensitivity and secretion, reduces glycaemia and improves liver function in NAFLD patients carrying the C282Y allele [67][68][69] [70]. Moreover, iron depletion is essential in the cellular response to hypoxic conditions, stabilizing the hypoxia-inducible factor (HIF)-1α, which is also involved in glucose uptake by inducing glucose transporter 1 (Glut1) and anaerobic glucose metabolism [71][72]. Dongiovanni and colleagues widely investigated the impact of iron depletion by deferoxamine on insulin signaling and glucose uptake in HepG2 cells and in rat livers. These authors have demonstrated that deferoxamine-induced iron depletion stabilizes HIF-1α and induces glucose uptake through Glut1, glucose utilization and insulin sensitivity [71].
Conversely, in C57Bl/6 wild-type male mice, dietary iron supplementation resulted in hypogonadism with a significant reduction of serum levels of testosterone [73], hyperglycemia, hyperinsulinemia, IR and hepatic iron accumulation. As a consequence of iron accumulation in adipocytes, iron-supplemented mice display lower adipose tissue mass and peripheral IR, testified by decreased phospho-Akt/Akt ratio and overexpression of adipokines including cytokine signaling-3 (Socs3), a target of hepcidin involved in IR [54].
Intriguingly, our unpublished data demonstrate that Hfe heterozygous (Hfe+/–) mice have a similar glucose concentration compared to wild-type (Hfe+/+) when they are fed a standard diet (94.8 ± 11 vs. 114 ± 41 mg/dL, respectively). However, after the administration of an iron-enriched diet for eight weeks, glucose levels were increased in both genotypes, but more so in the wild-type (157.8 ± 16 vs. 184.6 ± 27 mg/dL; p < 0.05). Moreover, fasting glucose levels and IR after insulin tolerance test (ITT) were higher in the wild-type compared to Hfe+/– mice treated with the same diet. Consistently, in wild-type mice, IR was paralleled by an impairment in insulin signaling activation and by a lower hepatic phospho-Akt/Akt ratio.
As we expected, hepatic iron and splenic contents were more severely increased in Hfe+/– mice compared to their wild-type littermates (700 ± 35 vs. 300 ± 15 μg/100 mg dry hepatic tissue and 450 ± 25 vs. 150 ± 10 μg/100 mg dry spleen tissue; p < 0.001), after the administration of the iron-enriched diet. However, the iron concentration in visceral adipose tissue was significantly enhanced in wild-type mice compared to Hfe+/– mice (10 ± 3 vs. 30 ± 5 μg/100 mg dry adipose tissue; p = 0.03), suggesting that adipose tissue iron overload may play a strong causative role in IR onset, more so than hepatic iron accumulation. Consistently, Hfe+/– mice fed an iron-enriched diet showed a decrease in adipose tissue resistin levels and an increase in adiponectin gene expression compared to their wild-type siblings fed the same diet, confirming that wild-type mice develop stronger peripheral IR than Hfe+/–. This effect seems to be mediated by the persistence of hepcidin presence in wild-type mice even after an iron challenge, as it occurs in DIOS patients differently from HH [74][75]. Indeed, DIOS and NAFLD disorders show elevated hepcidin levels related to hyperferritinemia. To confirm this hypothesis, we have recently demonstrated that iron challenge in iron-depleted DIOS patients did not restrain iron absorption despite adequate hepcidin production, suggesting that hepcidin resistance (i.e., the impaired hepcidin activity) and not the deficit of hormone production is involved in DIOS pathogenesis [76].
These novel findings pave the way to explore therapeutic strategies and clinical trials, based on hepcidin concentration control in order to limit iron overload in patients. Moreover, nutritional personalized therapeutic approaches aimed at reducing iron, carbohydrate and saturated fatty acid intake from diet may slow the raising of T2D and hepatic complications in HH patients.
2.4. Role of Bioactive Compounds in Iron Metabolism
Bioactive compounds are extra-nutritional substances naturally present in small quantities in foods that impact on metabolism at several levels. Indeed, they can influence intestinal transit, modify nutrient absorption and excretion and exert detoxifying and antioxidant actions [77].
Bioactive compounds affect dietary iron absorption by acting as inhibitors or enhancers of non-heme iron uptake [78]. Moreover, many bioactive compounds are phytochelators, i.e., plant products that have detoxifying properties and are able to prevent iron oxidative damage. Therefore, they can be successfully used (together with synthetic iron chelator drugs) in both primary iron overload disease, such as HH, and in secondary iron overload disease, namely β-Thalassemia [79].
The manipulation of diet composition should take into account the impact of these molecules on iron bioavailability to tailor a dietetic approach in subjects with perturbations of iron homeostasis. Indeed, due to their chelating and redox properties, some of these compounds reduce serum or tissue iron levels thereby their consumption should be recommended in patients with iron overload and avoided in those with iron deficiency. Moreover, bioactive compounds promoting iron mobilization or increasing iron uptake, which are beneficial for patients with iron deficiency, should be avoided in iron overloaded patients.
The next sections summarize the role of bioactive compounds in iron metabolism, focusing on their possible use in HH management and treatment.
2.4.1. Dietary Inhibitors of Iron Absorption: Phytates, Polyphenols, Calcium and Milk Soybean and Egg Proteins
2.4.1.1. Phytates
Phytic acid (myo-inositol-6-phosphate) is the main inhibitor of iron absorption. Phytates are largely present in all edible plant seeds (grains, legumes, oilseed and nuts) and to a lesser extent in roots, tubers and vegetables [80]. Due to their negative charge, in physiological conditions, phytates chelate multivalent cations including iron, zinc, magnesium and calcium [81]. The resulting complexes are soluble in the acidic environment of the stomach but precipitate in the small intestine where the pH reaches the neutral values, making iron (and other minerals) poorly absorbable. The phytate content in foods was found to be inversely related to the iron absorption [82]. Moreover, phytates exert their negative effects on iron absorption in a dose-dependent manner starting at very low concentrations (2–10 mg/meal) [83][84]. In a mouse model of iron overload, the chelating action of phytates attenuates iron-induced oxidative stress and alleviates liver injury [85]. Therefore, due to the iron lowering effect of these nutraceutical compounds, the introduction of nutrients rich in phytates could be considered a possible dietary intervention aimed to reduce iron bioavailability in HH patients who are predisposed to liver damage.
2.4.2. Polyphenols
Polyphenols are plant secondary metabolites, which contain one or more aromatic rings bearing one or more hydroxyl groups. They exert a pivotal role in the interactions between plants and the surrounding environment, as well as in plant reproduction and defense. Indeed, phenols are needed for the attraction of pollinators or seed-dispersing animals, for the protection against herbivores, microbe and viruses, ultraviolet radiation, oxidant agents and fluctuation of nutrients in soil [86]. Polyphenols are present in a wide variety of vegetarian food (vegetables, fruit, some cereals and legumes) and beverages (tea, coffee and wine). The phenolic compounds during digestion bind iron, likely through their galloyl groups, and polymerize it in insoluble complexes, which make iron unavailable for absorption [87].
In the past 40 years, iron-chelating properties of polyphenols have been explored [88] in several studies investigating their effects on iron absorption in humans. The consumption of yod kratin (a highly polyphenols-rich vegetable) reduced iron absorption from a composite meal by almost 90% in 83 Thai male healthy volunteers [89]. Polyphenols-rich beverages such as tea, coffee and cacao have been shown to lower iron absorption accordingly to the total quantity of polyphenols content [90] [91] [92] [93].
Interestingly, the consumption of polyphenols-containing beverages has been suggested as a useful strategy to reduce iron absorption in iron overload disorders. Indeed, Kaltwasser and colleagues demonstrated that iron absorption was significantly reduced when a meal was accompanied by tea instead of water in 18 HH patients. Moreover, regular tea drinking with meals reduced the frequency of phlebotomies required in the management of the diseases [94].
It has been shown that food supplementation with polyphenolic extract from tea and rosemary reduced iron absorption in 27 premenopausal women with normal iron stores, suggesting that increased flavonoids supplementations may maintain a relatively low iron status and may, therefore, reduce the risk of iron overload [95].
Among polyphenols, flavonoids (primarily quercetin) are considered the main dietary inhibitors of duodenal iron absorption. However, the exact molecular mechanisms through which flavonoids reduce iron bioavailability remain elusive. The majority of the evidence comes from studies on quercetin effects on non-heme iron absorption in vivo. It has been demonstrated in a rat model that quercetin chelates iron in the intestinal lumen and blocks it inside the enterocytes, by increasing iron apical uptake and preventing basolateral transport into the bloodstream [96], thus reducing intestinal iron absorption. Indeed, long-term administration of 50 mg/kg/daily for 10 days of quercetin by gavages decreases duodenal DMT1, duodenal cytochrome b (Dcytb) and Fpn mRNA levels and leads to animal iron depletion. Moreover, intraperitoneal short-term administration of 50 mg/kg of quercetin resulted in a significant reduction of iron parameters and upregulation of HAMP expression in the duodenum [97]. This evidence is intriguing and has a potential therapeutic translational relevance for patients with iron deficiency as well as for those with iron overload. Indeed, quercetin supplementation was successfully tested in 84 patients with β-Thalassemia, who usually develop secondary iron overload due to blood transfusion therapy. Administration of 500 mg/daily of quercetin after a meal significantly reduced serum iron levels, ferritin, transferrin saturation and inflammatory markers such as tumor necrosis factor-α (TNF-α) and high-sensitivity C-reactive protein) compared to both baseline and placebo groups suggesting that quercetin may reduce iron overload in beta-thalassemic patients by exerting its iron-chelating activity [98].
However, quercetin effects on iron overloaded patients have not been investigated yet. Nevertheless, other polyphenols that belong to the flavonoid family were tested with conflicting results. Preliminary data suggest that consumption of sylibin, another flavonoid derived from milk thistle, reduced postprandial iron serum levels in 10 C282Y homozygous patients [99]. Conversely, in vitro studies demonstrated that the iron-chelating ability of grape seed proanthocyanidin extract, a flavonoid of the tannin class, was equivalent to 30 mM of the iron chelator deferoxamine, in a 50 mM solution of ferrous sulfate [100]. In a double-blind randomized controlled study, the nutritional supplementation with procyanidins in HH patients did not significantly reduce dietary iron absorption during an iron-rich meal [101].
These conflicting results are possibly attributable to the fact that the effectiveness of polyphenols in chelating iron could be impaired by their intrinsic bioavailability, by the incorrect experimental timing, or by the different methods used to detect iron absorption. Therefore, further studies on a larger set of patients are required to clarify the role of polyphenols in iron absorption.
Nevertheless, polyphenols provide many health benefits and the consumption of polyphenol enriched foods is widespread. Consistently, we could speculate that chronic consumption of foods scarce in iron and rich in inhibitors of iron availability, such as in a vegan or vegetarian diet, might contribute to the diffusion of iron deficiency syndromes among determinate groups of individuals such as children, pregnant women or the elderly. Conversely, consumption of a polyphenol-enriched diet might be beneficial for individuals at risk of iron overload, such as HH patients.
2.4.3. Calcium
Calcium is the only dietary factor able to inhibit both heme- and non-heme iron absorption [102] in a dose-dependent fashion[103]. The mechanism underlying its inhibitory effect is not fully understood but it has been proposed that calcium may reduce the initial iron mucosal uptake by enterocytes without affecting its basolateral transfer [104]. Moreover, calcium has been shown to delay iron uptake in the intestinal epithelial cells in animal models [105]. In addition, an in vitro study suggested that calcium reduces iron bioavailability by decreasing DMT1 expression at the apical cell membrane, thereby downregulating iron transport into the cell [106].The question of whether the individual iron status may play a role in iron-calcium interaction has to be clearly addressed. Cook and colleagues described a significant inhibitory effect of calcium administered as calcium carbonate in 61 volunteers with low iron stores [102], the same treatment inhibited iron absorption in postmenopausal women with normal iron stores [107]. A recent review [108] of epidemiological studies that focused on the relationship between iron status and calcium intake indicates that calcium supplementation is unlikely to have significant effects on iron stores, although iron supplementation may be indicated during pregnancy to prevent anemia in women with high milk intake [109].
2.4.4. Proteins
Proteins deriving from soybean, milk and eggs have been shown to inhibit iron absorption [110]. Indeed, casein and whey, the two major bovine milk fractions, and egg white seem to prevent iron absorption in humans [111] [112] . Furthermore, an epidemiological study of the phenotypical expression of HFE C282Y, H63D and S65C mutations in 1294 Danish men demonstrated that high milk consumption decreased the clinical phenotypic expression of C282Y and H63D mutations by lowering serum ferritin levels [113].
Among proteins of a plant source, soybean derived ones have strong inhibitory effects on iron absorption [83]. However, when they were enzymatically digested by hydrolases to remove phytates from the matrix, the percentage of iron absorption increased by 19-fold suggesting that the iron absorption preventing the effect of soy is mediated by both phytates and proteins [114].
2.4.5. Dietary Enhancer of Iron Absorption: Vitamins and “Meat Factor”
2.4.5.1. Vitamins
Vitamins are essential micronutrients required for metabolism maintenance as they are cofactors for several enzymes. Vitamin A is a fat-soluble antioxidant molecule and it is also involved in iron homeostasis. It has been demonstrated that Vitamin A positively affects intestinal iron absorption. Indeed, its supplementation has been shown to prevent the inhibitory effect of phytates and polyphenols on iron absorption by sequestering iron in soluble complexes thus improving non-heme iron absorption from phytates-rich foods such as rice, wheat and corn [115].
Consistently, Vitamin A serum levels are positively related to iron indices (serum iron, hemoglobin and transferrin saturation) in humans [116] and its deficiency often co-occurs with iron deficiency anemia in some populations [117] [118]. Nevertheless, Vitamin A deficiency has been reported also in patients with idiopathic hemochromatosis [119]. Interestingly, this finding was confirmed in a more recent case-control study [120]. Indeed, C282Y homozygous patients showed lower Vitamin A serum levels than healthy controls at baseline. Remarkably, the reduction of oxidative stress and the mild anemic status induced by venesection therapy restored Vitamin A serum levels close to those of healthy subjects.
Moreover, Vitamin A deficiency has been associated with reduced iron incorporation in erythrocytes [121], alteration in red blood cell morphology [122] and mild anemia [123] but also with increased iron absorption and accumulation in the liver and spleen [124] in animal models.
The role of Vitamin A in the regulation of iron homeostasis is intricate and may involve several mechanisms. Experimental evidence showed that vitamin A deficiency is positively related to the upregulation of hepatic HAMP expression in vivo without affecting the expression of genes involved in iron absorption. Moreover, it induces FPN expression in vitro in a hepcidin-independent manner, thus suggesting that vitamin A deficiency promotes iron absorption by inducing its mobilization [125]. In addition, the exposure of Caco-2, an enterocyte cell line, to the Vitamin A precursor β-carotene reduced intracellular ferritin and in turn increased FPN expression leading to the release of intracellular trapped iron [126].
Vitamin C or ascorbic acid is the main enhancer of non-heme iron absorption. Vitamin C promotes iron absorption through different synergic mechanisms: (1) improving gastric iron solubility by acidifying the stomach content; (2) preventing the formation of ferric insoluble complexes by reducing iron from ferric to ferrous form thus making it available for transport by DMT1; (3) maintaining iron solubility in the duodenal neutral environment by chelating ferrous iron [127].
Several single-meal studies in human volunteers have shown that Vitamin C improves iron absorption both as a food source [128] [129] [130] [131] and as a supplement [130] [132]. Noteworthy, the positive effect of Vitamin C on iron absorption widely varies according to the timing of intake, the amount ingested and the meal composition. Indeed, increased iron absorption was not observed when Vitamin C was administered several hours before an iron-content meal [133]. In addition, 50 mg of Vitamin C increased iron absorption to 61% from a hamburger meal and to 164% from a pizza meal [130]. Meal composition, as well as the whole food matrix, impact the effectiveness of Vitamin C in modulating iron absorption. Although Vitamin C has been shown to overcome the negative effects of the inhibitors of iron absorption, including phytates [83], polyphenols [134], calcium and milk proteins [135], the high content in polyphenols and phytates of some fruits and vegetables may abolish the positive effect of Vitamin C [136] [137].
While the potentiating effects of vitamin C on iron absorption in single-meal studies have been convincingly demonstrated, the impact of vitamin C on iron status is not unknown. Limited available evidence deriving from long-term studies in subjects with low body iron stores are conflicting. Indeed, two studies showed a significant improvement of iron status in pre-school children with mainly plant-based diets [138] [139] and in young women with low iron stores who consumed 164 mg/day of Vitamin C for 16 weeks [140]. Conversely, Vitamin C supplementation (1500 mg/day for 5 weeks or 50 mg/day for 24 weeks) failed to improve serum ferritin in women with low body iron stores consuming western diets [141] [142].
However, further studies are required to assess the effectiveness of Vitamin C supplementation in improving iron status as differences in sample size, length of intervention, time and dose of supplementation and age of participants make these results difficult to define.
The impact of Vitamin C on iron stores in HH patients has not been investigated yet. However, Milward and colleagues showed retrospectively that non-citrus fruit consumption had significant protective effects on iron stores in HH male patients, independently of HFE genotype [48]. Although neither citrus fruit consumption nor Vitamin C supplementation increased iron stores, the consumption of 14 or more pieces of non-citrus fruit per week resulted in a 20% reduction of ferritin serum levels, suggesting that a diet rich in non-citrus fruits should be recommended in HH patients whose iron levels are not well controlled by phlebotomy or blood donations. Thus, it could represent a helpful strategy to reduce the frequency of phlebotomy required to reach iron depletion in HH patients.
2.4.6. Muscle Tissues from Meat, Fish and Poultry: The “Meat Factor” Effect
An animal-based diet has an important role in iron balance not only for its own iron content but also for the ability of animal foodstuffs (mainly meat, fish and poultry) to enhance non-heme iron absorption [143]. Indeed, in their systematic review, Jackson and colleagues suggested a positive correlation between the intake of animal flesh foods and increased iron stores in healthy adults within developed countries [144]. In addition, several single-meal studies showed that muscle tissues from meat, fish and poultry have a positive effect on iron absorption from vegetarian meals [145], to the same extent as Vitamin C [146]. Indeed, addition of chicken beef or fish to maize meal increased iron absorption by 2–3 fold [147]. To date, the nature of the “meat factor” remains elusive and the mechanisms underlying the enhancing effect of muscle tissues are still debated. Zang and colleagues suggested that muscle tissues, defined as a “meat factor”, may enhance non-heme iron absorption by promoting iron solubility in the stomach (where it stimulates gastric acid secretion) and by maintaining it (both by stimulating gastrin and/or other gastric factors and by chelating non-heme iron) during digestion in neutral pH conditions of the duodenum thus making non-heme iron readily absorbable [148]. More recent evidence suggests that the “meat factor” enhancing effect on iron absorption could be attributable to the ability to reduce and chelate iron of Cysteine-containing peptides derived from myofibrillar protein digestion, glycosaminoglycans and L-α-glycerophosphocholine [149] [150] [151].
2.4.7. Prevention of Detrimental Effects of Iron-Induced Oxidative Stress: Antioxidants
2.4.7.1. Vitamin E
Vitamin E (RRR-alpha-tocopherol) is an important lipid-soluble antioxidant compound. It acts as a scavenger of free radicals protecting biological membranes from radical-mediated injury [152] [153]. The liver is involved in Vitamin E metabolism. Indeed, after duodenal uptake, Vitamin E is mainly transferred as chylomicrons to hepatic parenchymal cells where it is re-oxidized and secreted with very low-density lipoprotein (VLDL) into the bloodstream [154].
Lipid peroxidation, due to iron-mediated oxidative stress, plays a pivotal role in the pathogenesis of liver damage in iron overload conditions [155]. Indeed, it results in a loss of integrity and impaired function of cellular organelles in experimental iron overload models [156] [157] [158]. Interestingly, lipid peroxidation was increased and antioxidant potential was impaired in HH patients [159].
Vitamin E supplementation exerts a protective effect on redox damage both in vivo and in vitro [160] [161]. Conversely, Vitamin E deficiency increases lipid peroxidation and promotes oxidant damage in hepatocytes [162] [163]. Moreover, iron overload has been shown to reduce Vitamin E levels in animal models [164]. Consistently, patients with HH have lower Vitamin E serum levels than healthy controls [165] [166]. Interestingly, in HH patients who have reached iron depletion by phlebotomy, serum levels of Vitamin E were comparable to those of controls, suggesting that the impairment of antioxidant potential due to Vitamin E deficiency may have a role in the pathogenesis of HH [166]. Therefore, it is reasonable to assume that Vitamin E supplementation in these patients may have beneficial effects on iron overload-induced redox injury by preventing tissue damage.
The positive effects of Vitamin E supplementation in preventing lipid peroxidation induced by iron free-radicals were successfully confirmed in animal models of dietary iron overload whereas they remain to be deeper investigated in HH patients [167] [168].
2.4.8. Phenolic Antioxidants: Flavonoids, Ferulic acid and Resveratrol
Among polyphenols, Flavonoids represent the major class of antioxidants. A flavonoid-rich extract of citrus fruits (orange and bergamot) has been shown to chelate iron and to reduce ROS production and membrane lipid oxidation by activating catalase enzymes in lung epithelial cell lines [169].
Similarly, grape seed extract (GSE) and epigallocatechin-3-gallate (EGCG), a tea-derived catechin, have potent antioxidant properties. Indeed, EGCG prevents oxidative stress by activating nuclear factor erythroid 2 related factor 2 (Nrf2) [170], a master transcriptional regulator of antioxidant genes [171]. Moreover, EGCG prevents intestinal iron absorption by reducing basolateral iron export in Caco-2 cells [172] and it may improve iron status and restore the redox potential in patients with HH. GSE-derived anthocyanins have been shown to antagonize oxidative injury by increasing total antioxidant potential and decreasing iron content through downregulation of Hamp and upregulation of Fpn expression in human embryo-derived hepatic cells [173]. Curcumin decreases iron levels in bone marrow, the spleen and the liver. In a mouse model of diet-induced subclinical iron deficiency, curcumin administration resulted in a dose-dependent reduction of hematocrit, hemoglobin, serum iron and transferrin saturation, exacerbating iron deficiency anemia. Consistently with the iron depletion status of these animals, curcumin treatment induced the activation of hepatic IRP and TfR-1 as well as the reduction of hepatic ferritin and HAMP. In vitro in HepG2 cells, curcumin treatment reduced HAMP expression and induced ferroportin thus leading to iron efflux from cells [174].
Ferulic acid is a phenolic compound abundantly present in grains, artichokes, coffee and fruits. Its antioxidant effect is mediated by the neutralization of free radicals [175]. Iron overloaded mice treated with sodium ferulate showed reduced hepatic iron-induced oxidative stress, which led to improving the mitochondrial membrane potential, reversing mitochondrial swelling and decreasing the production of ROS, therefore, reducing liver injury [176]. However, the effects of ferulic acid on iron-induced oxidative stress have not yet been investigated in HH patients.
Resveratrol (3,5,4-trans-trihydroxystilbene) is a compound that belongs to the stilbene subclass of polyphenols. Resveratrol has pleiotropic effects and acts as an antioxidant, anti-inflammatory and anti-apoptotic [177]. In murine models of genetic HH exposed to chronic iron-overload, resveratrol reduced iron-induced hepatosplenomegaly, oxidative stress, hepatic fibrosis and inflammation and a pro-apoptotic status. Resveratrol supplementation provided protection from iron-mediated tissue injury in these models, whereas it did not affect the degree of hepatic iron overload. Indeed, by restoring sirtuin 1 (SIRT1) expression, resveratrol induced forkhead box O1 (FOXO1) deacetylation and activated FOXO1-dependent anti-oxidant response during oxidative stress [178]. Therefore, resveratrol supplementation may represent a potential therapy for iron-induced liver damage in patients with HH.
The main dietary bioactive compounds and their impact on iron metabolism are listed in Table 1.
Table 1. Dietary bioactive molecules and their impact on iron metabolism.
| Molecules | Source | Action on Iron Absorption | Mechanism of Action | Evidence in HH Patients or Animal Models | Ref |
|---|---|---|---|---|---|
| Vitamin C | Mineral | Antioxidant/enhancer | Redox/chelation | Reduction of iron stores | [48] |
| Phytates | Plants | Inhibition | Chelation | Improvement of iron-induced oxidative stress and liver injury in animal models | [80] |
| Polyphenols | Plants | Inhibition | Chelation | Reduction of iron absorption and of the frequency of phlebotomies | [94] [99] |
| Calcium | Mineral | Inhibition | Reducing iron uptake | Not investigated | |
| Soybean, milk and egg proteins | Animal | Inhibition | Unknown | Reduction of serum ferritin | [114] |
| Vitamin A | Mineral | Enhancer | Hepcidin-dependent upregulation of hepatic Hamp and duodenal Fpn | Vitamin A deficiency reported in HH | [119] [120] |
| Vitamin E | Mineral | Antioxidant | Scavenge ROS protecting membranes from lipid peroxidation | Vitamin E deficiency reported in HH | [160] [161] [162] [163] [164] [165] [166] [167] [168] |
| “Meat factor” | Animal | Enhancer | Chelation | Not investigated | |
| Orange/bergamot flavonoid-rich extracts | Plants | Antioxidant | Chelation of iron and reduction of ROS | Not investigated | [169] |
| EGCG | Plants | Antioxidant/inhibitor | Chelation, reduction of basolateral iron export and activation of Nrf2 in Caco-2 and in human mesenchymal stem cells | Not investigated | [170][171][172] |
| GSE | Plants | Antioxidant/inhibitor | Chelation, reduction of basolateral iron export in Caco-2 | Not investigated | [173] |
| Curcumin | Plants | Antioxidant | Reduction of iron content of liver, spleen and bone marrow; activation TfR-1 and IRP, repression of hepatic ferritin and hepcidin synthesis | Not investigated | [174] |
| Ferulic acid | Plants | Antioxidant | Reduction of liver damage by increasing hepatic antioxidants and mitochondrial membrane potential | Not investigated | [175][176] |
| Resveratrol | Plants | Antioxidant | Upregulation of SIRT1 expression and activation of FOXO1-dependent anti-oxidant response | Not investigated | [177][178] |
2.5. Epigenetic Modifiers of HFE: miRNAs-Nutrients Interaction in the Modulation of HH Phenotype
Epigenetic modifiers in HH can represent novel molecular predictors that can determine not only the early risk assessment but also the disease progression and prognosis, providing a new perspective on HH management. Indeed, the phenotypic discrepancies of HH manifestations may also be the consequence of the complex gene–environment interplay, explained by epigenetic mechanisms, hereditable but reversible modifications that modulate the transcriptome in response to environmental cues, without altering DNA sequence [179]. Epigenetics embraces a broad number of events such as alterations of DNA nucleotides (i.e., methylation of CpG dinucleotides, known as CpG islands), modifications of histones and regulation of transcription by altering mRNA stability through small RNA molecules such as microRNAs (miRNAs). Specifically, miRNAs are short non-protein coding, single-strand RNAs of 19–22 nucleotides that regulate gene expression and cell-to-cell communications as it occurs between hepatocytes and Kupffer cells or hepatic stellate cells (HSCs) during hepatic inflammation and fibrogenesis [179]. Indeed, miRNAs can target mRNAs through complementary base-pairing, thereby leading to the post-transcriptional repression of targeted protein-coding genes [180] [181] [182].
Dietary habits may strongly affect the hepatic epigenetic landscape, introducing, for example, modifications of cellular homeostasis and macro/micronutrient metabolism thus influencing the health status but also affect pathological conditions [183] [184]. More than two thousand miRNAs have been described [185]. Evidence has revealed that they are encoded in a wide variety of intronic, exonic or intergenic sequences and that they directly target up to 60% of all human genes [185]. In particular, miRNAs may exert either the silencing of the target mRNAs or the repression of protein synthesis through different mechanisms [180]. Therefore, miRNAs may regulate at multiple levels iron metabolism by targeting the majority of genes involved in iron uptake, storage and recycling at cellular and systemic level [186]. Likewise, each miRNA may target multiple genes (multi-functionality) or multiple miRNAs can target a single gene (redundancy), supporting the notion that miRNAs may have an extensive regulatory ability in iron homeostasis and a strikingly diverse effect on health status and disease [180]. In particular, heme plays a crucial regulatory role in miRNA biogenesis, through the direct interaction with DiGeorge syndrome critical region 8 (DGCR8), which participates in precursor-miRNA cleavages [187]. As a consequence, during heme deficiency, the maturation of miRNA precursors is predicted to be slow, resulting in a global decrease of mature miRNA abundance.
Moreover, several miRNAs among which miR-22, miR-200a, and miR-320 are predicted to bind the 3′UTR sequence of TfR-1, essential for iron uptake from the blood. Specifically, the overexpression of miR-320 hampers TfR-1 expression and subsequently lowers iron availability and cell proliferation [188]. Nevertheless, miR-15a/b, miR-223 and Let-7d are involved in the transferrin cycle, interfering with the iron release from the endosome via DMT1. The iron retention in endosomes may indicate functional cellular iron deficiency [186]. Lactoferrin, which is another member of transferrin family, along with its receptor has been characterized as a functional target of miR-214 in both HC11 and MCF7 cancer cells [189]. The overexpression of miR-214 markedly decreased lactoferrin expression, also reducing apoptotic processes [189]. The post-transcriptional expression of the lactoferrin receptor, mainly localized in the apical membrane of enterocytes, is mediated also by miR-584 in both Caco-2 cells and in the mouse small intestine during the perinatal period, thus participating in the absorption of lactoferrin-bound iron from breast milk [190]. Finally, miR-200b has been well known to regulate iron cellular storage mediating ferritin regulation [191].
An example of miRNAs that are well established to be closely entangled with iron metabolism is represented by miR-122. Indeed, miR-122, the most abundant miRNA in the liver, is reduced in genetically-induced murine models of HH and accordingly even in hepatic biopsies of C282Y homozygous patients. Consistently, the inhibition of miR-122 in wild-type mice through intraperitoneal injection of antagomiR, hesitates in hampered circulating and tissue iron concentrations in spleen and liver, mildly impairing hematopoiesis [192]. Moreover, miR-122 deficiency enhances the expression of Hfe, hemojuvelin (Hjv) and Hamp, which are involved in the sensing of systemic iron levels [192]. miRNA-target prediction analyses, indeed, reveal the presence of miR-122 binding sites on Hfe, Hjv and Hamp 3′ UTR regulatory sequences. These observations have been confirmed in primary hepatocytes, after the transfection of miR-122 mimics or inhibitors, further corroborating that miR-122 is enabled to modulate systemic iron utilization and may be implicated in the prevention of iron overload and in iron overload-related complications, such as cirrhosis and hepatocellular carcinoma [186].
Among the bioactive compounds, several polyphenols i.e., quercetin, coffee polyphenols and grape seed proanthocyanidin may directly influence miR-122 expression, affecting, in turn, different metabolic pathways such as de novo lipogenesis, ameliorating body fat deposition and preventing diet-induced hepatic abnormalities in murine models [193][194]. The modulation of iron absorption by polyphenol-related miRNAs has been more clearly investigated [102]. Lesjak M. and collaborators demonstrate that quercetin, the most abundant dietary polyphenol and potent iron chelator, is able to downregulate Fpn levels both in in vivo and in vitro models. Indeed, the downregulation of Fpn has been attributable to miR-17–3p and its interaction with Fpn 3′UTR sequence. This data sustains the idea that dietary polyphenols severely impact on iron bioavailability thereby limiting the rate of intestinal iron absorption[96].
However, the field of diet-epigenome interaction in HH and its comorbidities remains largely unexplored. Overall, this evidence supports the hypothesis that a polyphenol-supplemented dietary approach to HH patients may improve tissue iron accumulation and eventually ameliorate liver injuries induced by iron depots, whereby modulating miRNA expression. This concept may provide a new example of gene-environment interaction that should be deeply investigated in the future.
An overview of the impact of bioactive compounds and dietary factors on iron metabolism is schematically represented in Figure 2.
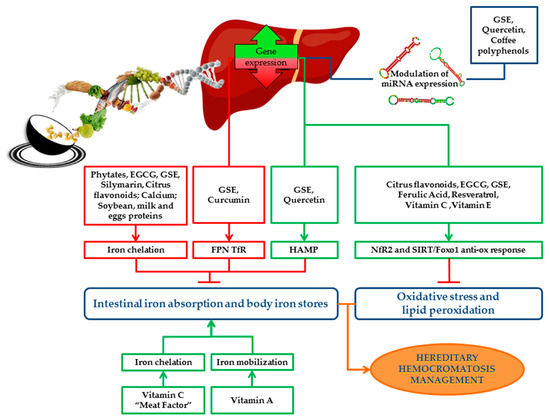
Figure 2. Schematic representation of the impact of bioactive compounds and dietary factors on iron metabolism. Dietary factors and bioactive compounds regulate iron metabolism directly, by enhancing/inhibiting its absorption, storage and recycling, or indirectly by the modulation of miRNAs, which regulate the expression of iron genes. Bioactive compounds act as antioxidants and protect cells and tissues from detrimental effects of iron overload by reducing oxidative stress and lipid peroxidation. The picture shows the main pathways induced (green boxes) or downregulated (red boxes) by bioactive compounds and the final effect on iron absorption, body iron stores, oxidative stress and lipid peroxidation (green arrows indicate positive effects; red “T” arrows indicate inhibitory ones). Nutritional interventions aimed at reducing iron absorption and improving iron-induced oxidative stress may be useful and safe strategies supporting phlebotomy in the management and treatment of HH patients. EGCG: Epigallocatechin-3-gallate; GSE: Grape seed extract; FPN: Ferroportin; TfR: Transferrin receptor; HAMP: Hepcidin antimicrobial peptide gene; ROS: Reactive oxygen species; Nrf2: Nuclear factor erythroid 2 related factor 2; SIRT1: Sirtuin 1; FOXO1: Forkhead box O1.
References
- Antonello Pietrangelo; Hereditary Hemochromatosis: Pathogenesis, Diagnosis, and Treatment. Gastroenterology 2010, 139, 393-408.e2, 10.1053/j.gastro.2010.06.013.
- Tomas Ganz; Systemic Iron Homeostasis. Physiological Reviews 2013, 93, 1721-1741, 10.1152/physrev.00008.2013.
- Brissot, P.; Pietrangelo, A.; Adams, P.C.; de Graaff, B.; McLaren, C.E.; Loreal, O.; Haemochromatosis. Nat. Rev. Dis. Primers 2018, 4, 18016, 10.1038/nrdp.2018.16.
- Luisa Salter-Cid; Anders Brunmark; Yuanhao Li; Didier Leturcq; Per A. Peterson; Michael R. Jackson; Young Yang; Transferrin receptor is negatively modulated by the hemochromatosis protein HFE: Implications for cellular iron homeostasis. Proceedings of the National Academy of Sciences 1999, 96, 5434-5439, 10.1073/pnas.96.10.5434.
- Alberto Piperno; Sara Pelucchi; Raffaella Mariani; Inherited iron overload disorders.. Translational Gastroenterology and Hepatology 2020, 5, 25-25, 10.21037/tgh.2019.11.15.
- Robert E. Fleming; Robert S. Britton; Iron Imports. VI. HFE and regulation of intestinal iron absorption. American Journal of Physiology-Gastrointestinal and Liver Physiology 2006, 290, G590-G594, 10.1152/ajpgi.00486.2005.
- John N. Feder; Andreas Gnirke; W. Thomas; Z. Tsuchihashi; D.A. Ruddy; A. Basava; F. Dormishian; R. Domingo; M.C. Ellis; A. Fullan; et al.L.M. HintonN.L. JonesB.E. KimmelG.S. KronmalP. LauerV.K. LeeD.B. LoebF.A. MapaE. McClellandN.C. MeyerG.A. MintierN. MoellerT. MooreE. MorikangC.E. PrassL. QuintanaS.M. StarnesR.C. SchatzmanK.J. BrunkeD.T. DraynaN.J. RischB.R. BaconR.K. Wolff A novel MHC class I–like gene is mutated in patients with hereditary haemochromatosis. Nature Genetics 1996, 13, 399-408, 10.1038/ng0896-399.
- P Holmström; J Marmur; G Eggertsen; M Gåfvels; Per Stal; Mild iron overload in patients carrying the HFE S65C gene mutation: a retrospective study in patients with suspected iron overload and healthy controls. Gut 2002, 51, 723-730, 10.1136/gut.51.5.723.
- Catherine Mura; Gérald Le Gac; Odile Raguénès; Anne-Yvonne Mercier; Alain Le Guen; Claude Férec; Relation between HFE Mutations and Mild Iron-Overload Expression. Molecular Genetics and Metabolism 2000, 69, 295-301, 10.1006/mgme.2000.2981.
- Carles De Diego; Maria José Murga; Pedro Martínez-Castro; Frequency ofHFEH63D, S65C, and C282Y Mutations in Patients with Iron Overload and Controls from Toledo, Spain. Genetic Testing 2004, 8, 263-267, 10.1089/gte.2004.8.263.
- Adams, P.C.; Reboussin, D.M.; Barton, J.C.; McLaren, C.E.; Eckfeldt, J.H.; McLaren, G.D.; Dawkins, F.W.; Acton, R.T.; Harris, E.L.; Gordeuk, V.R; et al.Leiendecker-Foster, C.Speechley, M.Snively, B.M.Holup, J.L.Thomson, E.Sholinsky, P.Hemochromatosis and Iron Overload Screening (HEIRS) Study Research Investigators Hemochromatosis and iron-overload screening in a racially diverse population. New England Journal of Medicine 2005, 352, 1769-1778, 10.1056/NEJMoa041534.
- Merryweather-Clarke, A.T.; Pointon, J.J.; Jouanolle, A.M.; Rochette, J.; Robson, K.J.; Geography of HFE C282Y and H63D mutations. . Genet. Test. 2000, 4, 183-198, 10.1089/10906570050114902.
- Raha-Chowdhury, R.G.J. . Localization, allelic heterogeneity, and origins of the hemochromatosis gene. In Hemochromatosis: Genetics, Pathophysiology, Diagnosis and Treatment;; Barton, J.C.E.C., Ed, Eds.; Cambridge University Press: Cambridge, 2000; pp. 75-95.
- Distante, S.; Robson, K.J.; Graham-Campbell, J.; Arnaiz-Villena, A.; Brissot, P.; Worwood, M.; The origin and spread of the HFE-C282Y haemochromatosis mutation.. Hum. Genet. 2004, 115, 269–279, 10.1007/s00439-004-1152-4.
- Remko S. Kuipers; Martine F. Luxwolda; D. A. Janneke Dijck-Brouwer; S. Boyd Eaton; Michael A. Crawford; Loren Cordain; Frits A. J. Muskiet; Estimated macronutrient and fatty acid intakes from an East African Paleolithic diet. British Journal of Nutrition 2010, 104, 1666-1687, 10.1017/s0007114510002679.
- Loren Cordain; S B Eaton; J Brand Miller; N Mann; K Hill; The paradoxical nature of hunter-gatherer diets: meat-based, yet non-atherogenic. European Journal of Clinical Nutrition 2002, 56, S42-S52, 10.1038/sj.ejcn.1601353.
- Richard F. Hurrell; Influence of Vegetable Protein Sources on Trace Element and Mineral Bioavailability. The Journal of Nutrition 2003, 133, 2973S-2977S, 10.1093/jn/133.9.2973s.
- Naugler, C; Hemochromatosis: A Neolithic adaptation to cereal grain diets.. Med. Hypotheses 2008, 70, 691-692, 10.1016/j.mehy.2007.06.020.
- Pennington, J.A.; Wilson, D.B.; Young, B.E.; Johnson, R.D.; Vanderveen, J.E.; Mineral content of market samples of fluid whole milk.. J. Am. Diet. Assoc 1987, 87, 1036-1042.
- Ekhard E. Ziegler; Consumption of cow's milk as a cause of iron deficiency in infants and toddlers. Nutrition Reviews 2011, 69, S37-S42, 10.1111/j.1753-4887.2011.00431.x.
- Jean-Pierre Bocquet-Appel; When the World's Population Took Off: The Springboard of the Neolithic Demographic Transition. Science 2011, 333, 560-561, 10.1126/science.1208880.
- Samuli Helle; Jon E. Brommer; Jenni E. Pettay; Virpi Lummaa; Matti Enbuske; Jukka Jokela; Evolutionary demography of agricultural expansion in preindustrial northern Finland. Proceedings of the Royal Society B: Biological Sciences 2014, 281, 20141559-20141559, 10.1098/rspb.2014.1559.
- Srdjan Denic; Mukesh M. Agarwal; Nutritional iron deficiency: an evolutionary perspective. Nutrition 2007, 23, 603-614, 10.1016/j.nut.2007.05.002.
- M. A. V. Bokhoven; C. T. B. M. V. Deursen; D. W. Swinkels; Diagnosis and management of hereditary haemochromatosis. BMJ 2011, 342, c7251-c7251, 10.1136/bmj.c7251.
- Beutler, E.; Felitti, V.J.; Koziol, J.A.; Ho, N.J.; Gelbart, T.; Penetrance of 845G--> A (C282Y) HFE hereditary haemochromatosis mutation in the USA.. Lancet 2002, 359, 211-218, 10.1016/S0140-6736(02)07447-0.
- Katrina J. Allen; Lyle Gurrin; Clare C. Constantine; Nicholas J. Osborne; Martin B. Delatycki; Amanda Nicoll; Christine E. McLaren; Melanie Bahlo; Amy Nisselle; Chris Vulpe; et al.Gregory J. AndersonMelissa C. SoutheyGraham G. GilesDallas R. EnglishJohn L. HopperJohn K. OlynykLawrie W. PowellRota M. Gertig Iron-Overload–Related Disease inHFEHereditary Hemochromatosis. New England Journal of Medicine 2008, 358, 221-230, 10.1056/nejmoa073286.
- Linda M. Fletcher; Jeannette L. Dixon; David M. Purdie; Lawrie W. Powell; Darrell H.G. Crawford; Excess alcohol greatly increases the prevalence of cirrhosis in hereditary hemochromatosis. Gastroenterology 2002, 122, 281-289, 10.1053/gast.2002.30992.
- Daniel F. Wallace; V. Nathan Subramaniam; Co-factors in liver disease: The role of HFE-related hereditary hemochromatosis and iron. Biochimica et Biophysica Acta (BBA) - General Subjects 2009, 1790, 663-670, 10.1016/j.bbagen.2008.09.002.
- Virginie Scotet; Marie-Christine Mérour; Anne-Yvonne Mercier; Brigitte Chanu; Thérèse Le Faou; Odile Raguénes; Gérald Le Gac; Catherine Mura; Jean-Baptiste Nousbaum; C. Férec; et al. Hereditary hemochromatosis: effect of excessive alcohol consumption on disease expression in patients homozygous for the C282Y mutation.. American Journal of Epidemiology 2003, 158, 129-134, 10.1093/aje/kwg123.
- Loreal, O.; Deugnier, Y.; Moirand, R.; Lauvin, L.; Guyader, D.; Jouanolle, H.; Turlin, B.; Lescoat, G.; Brissot, P.; . Liver fibrosis in genetic hemochromatosis. Respective roles of iron and non-iron-related factors in 127 homozygous patients. . J. Hepatol. 1992, 16, 122–127, 10.1016/s0168-8278(05)80104-7.
- P C Adams; S Agnew; Alcoholism in hereditary hemochromatosis revisited: Prevalence and clinical consequences among homozygous siblings. Hepatology 1996, 23, 724-727, 10.1002/hep.510230411.
- Duygu Dee Harrison-Findik; Denise Schafer; Elizabeth Klein; Nikolai A. Timchenko; Hasan Kulaksiz; Dahn Clemens; Evelyn Fein; Billy Andriopoulos; Kostas Pantopoulos; John Gollan; et al. Alcohol Metabolism-mediated Oxidative Stress Down-regulates Hepcidin Transcription and Leads to Increased Duodenal Iron Transporter Expression. Journal of Biological Chemistry 2006, 281, 22974-22982, 10.1074/jbc.m602098200.
- Duane, P.; Raja, K.B.; Simpson, R.J.; Peters, T.J.; Intestinal iron absorption in chronic alcoholics. . Alcohol Alcohol. 1992, 27, 539-544.
- Yutaka Kohgo; Katsuya Ikuta; Takaaki Ohtake; Yoshihiro Torimoto; Junji Kato; Iron overload and cofactors with special reference to alcohol, hepatitis C virus infection and steatosis/insulin resistance. World Journal of Gastroenterology 2007, 13, 4699-4706, 10.3748/wjg.v13.i35.4699.
- Bridle, K.; Cheung, T.K.; Murphy, T.; Walters, M.; Anderson, G.; Crawford, D.G.; Fletcher, L.M.; Hepcidin is down-regulated in alcoholic liver injury: Implications for the pathogenesis of alcoholic liver disease.. Alcohol. Clin. Exp. Res. 2006, 30, 106–112, 10.1111/j.1530-0277.2006.00002.x.
- Duygu Dee Harrison-Findik; Elizabeth Klein; Callie Crist; John Evans; Nikolai Timchenko; John Gollan; Iron-mediated regulation of liver hepcidin expression in rats and mice is abolished by alcohol. Hepatology 2007, 46, 1979-1985, 10.1002/hep.21895.
- Michael Zimmermann; Nourredine Chaouki; Richard F Hurrell; Iron deficiency due to consumption of a habitual diet low in bioavailable iron: a longitudinal cohort study in Moroccan children. The American Journal of Clinical Nutrition 2005, 81, 115-121, 10.1093/ajcn/81.1.115.
- Michael Zimmermann; Richard Hurrell; Nutritional iron deficiency. The Lancet 2007, 370, 511-520, 10.1016/s0140-6736(07)61235-5.
- Nancy Andrews; Disorders of Iron Metabolism. New England Journal of Medicine 1999, 341, 1986-1995, 10.1056/nejm199912233412607.
- Roy, C.N.; Enns, C.A.; Iron homeostasis: New tales from the crypt. . Blood 2000, 96, 4020–4027.
- Charles E. Carpenter; Arthur W. Mahoney; Contributions of heme and nonheme iron to human nutrition. Critical Reviews in Food Science and Nutrition 1992, 31, 333-367, 10.1080/10408399209527576.
- Diego Moretti; Gerrigje M Van Doorn; Rine W Swinkels; Alida Melse-Boonstra; Relevance of dietary iron intake and bioavailability in the management of HFE hemochromatosis: a systematic review. The American Journal of Clinical Nutrition 2013, 98, 468-479, 10.3945/ajcn.112.048264.
- W. R. Bezwoda; P. B. Disler; S. R. Lynch; R. W. Charlton; J. D. Torrance; D. Derman; T. H. Bothwell; R. B. Walker; F. Mayet; Patterns of Food Iron Absorption in Iron-Deficient White and Indian Subjects and in Venesected Haemochromatotic Patients. British Journal of Haematology 1976, 33, 425-436, 10.1111/j.1365-2141.1976.tb03560.x.
- Bezwoda, W.R.; Bothwell, T.H.; Derman, D.P.; MacPhail, A.P.; Torrance, J.D.; Charlton, R.W.; Effect of diet on the rate of iron accumulation in idiopathic haemochromatosis. . S. Afr. Med. J. 1981, 59, 219–222..
- V R Gordeuk; Laura Lovato; James C Barton; Mara Vitolins; Gordon McLaren; Ronald T Acton; Emily L. Harris; Mark Speechley; John H Eckfeldt; Sharmin Diaz; et al.Phyliss SholinskyP. C. Adams Dietary Iron Intake and Serum Ferritin Concentration in 213 Patients Homozygous for the HFE C282Y Hemochromatosis Mutation. Canadian Journal of Gastroenterology 2012, 26, 345-349, 10.1155/2012/676824.
- D C. Greenwood; Janet E. Cade; J A. Moreton; B O??hara; Victoria Burley; Juliette Randerson-Moor; K Kukalizch; D Thompson; M Worwood; D. Timothy Bishop; et al. HFE Genotype Modifies the Influence of Heme Iron Intake on Iron Status. Epidemiology 2005, 16, 802-805, 10.1097/01.ede.0000181306.85583.ea.
- Peeters, P.H.; Grobbee, D.E.; Roest, M.; Voorbij, H.A.; van der Schouw, Y.T.; HFE genotypes and dietary heme iron: No evidence of strong gene-nutrient interaction on serum ferritin concentrations in middle-aged women. . Nutr. Metab. Cardiovasc. Dis. NMCD 2006, 16, 60–68, 10.1016/j.numecd.2005.07.008.
- Elizabeth A. Milward; Surinder K. Baines; Matthew Knuiman; Helen C. Bartholomew; Mark L. Divitini; David G. Ravine; David Bruce; John K. Olynyk; Noncitrus Fruits as Novel Dietary Environmental Modifiers of Iron Stores in People With or Without HFE Gene Mutations. Mayo Clinic Proceedings 2008, 83, 543-549, 10.4065/83.5.543.
- Silvia Fargion; Michela Mattioli; Anna Ludovica Fracanzani; Maurizio Sampietro; Dario Tavazzi; Paolo Fociani; Emanuela Taioli; Luca Valenti; Gemino Fiorelli; Hyperferritinemia, iron overload, and multiple metabolic alterations identify patients at risk for nonalcoholic steatohepatitis. The American Journal of Gastroenterology 2001, 96, 2448-2455, 10.1111/j.1572-0241.2001.04052.x.
- José-Manuel Fernández-Real; Wifredo Ricart-Engel; Enric Arroyo; Rafael Balançá; Roser Casamitjana-Abella; Dolores Cabrero; Miquel Fernández-Castañer; Joan Soler; Serum ferritin as a component of the insulin resistance syndrome.. Diabetes Care 1998, 21, 62-68, 10.2337/diacare.21.1.62.
- John D. Ryan; Andrew E. Armitage; Jeremy F. Cobbold; Rajarshi Banerjee; Oscar Borsani; Paola Dongiovanni; Stefan Neubauer; Reza Morovat; Lai Mun Wang; Sant-Rayn Pasricha; et al.Silvia FargionJane CollierEleanor BarnesHal DrakesmithLuca ValentiMichael Pavlides Hepatic iron is the major determinant of serum ferritin in NAFLD patients. Liver International 2017, 38, 164-173, 10.1111/liv.13513.
- Giulio Marchesini; Mara Brizi; Giampaolo Bianchi; Sara Tomassetti; Elisabetta Bugianesi; Marco Lenzi; Arthur J. McCullough; Stefania Natale; Gabriele Forlani; Nazario Melchionda; et al. Nonalcoholic fatty liver disease: a feature of the metabolic syndrome.. Diabetes 2001, 50, 1844-1850, 10.2337/diabetes.50.8.1844.
- Luca Valenti; P Dongiovanni; Anna Ludovica Fracanzani; G Santorelli; E Fatta; C Bertelli; E Taioli; G Fiorelli; Silvia Fargion; Increased susceptibility to nonalcoholic fatty liver disease in heterozygotes for the mutation responsible for hereditary hemochromatosis.. Digestive and Liver Disease 2003, 35, 172-178, 10.1016/s1590-8658(03)00025-2.
- Paola Dongiovanni; Massimiliano Ruscica; Raffaela Rametta; Stefania Recalcati; Liliana Steffani; Stefano Gatti; Domenico Girelli; Gaetano Cairo; Paolo Magni; Silvia Fargion; et al.Luca Valenti Dietary Iron Overload Induces Visceral Adipose Tissue Insulin Resistance. The American Journal of Pathology 2013, 182, 2254-2263, 10.1016/j.ajpath.2013.02.019.
- Larry W. Oberley; Free radicals and diabetes. Free Radical Biology and Medicine 1988, 5, 113-124, 10.1016/0891-5849(88)90036-6.
- Paola Dongiovanni; Claudia Lanti; Stefano Gatti; Raffaela Rametta; Stefania Recalcati; Marco Maggioni; Anna Ludovica Fracanzani; Patrizia Riso; Gaetano Cairo; Silvia Fargion; et al.Luca Valenti High Fat Diet Subverts Hepatocellular Iron Uptake Determining Dysmetabolic Iron Overload. PLOS ONE 2015, 10, e0116855, 10.1371/journal.pone.0116855.
- Witte, D.L.; Crosby, W.H.; Edwards, C.Q.; Fairbanks, V.F.; Mitros, F.A.; Practice guideline development task force of the College of American Pathologists. Hereditary hemochromatosis. . Clin. Chim. Acta Int. J. Clin. Chem. 1996, 245, 139–200, 10.1016/0009-8981(95)06212-2.
- Tomi-Pekka Tuomainen; Kristiina Nyyssönen; Riitita Salonen; Arja Tervahauta; Heikki Korpela; Timo A. Lakka; George A Kaplan; Jukka T. Salonen; Body Iron Stores Are Associated With Serum Insulin and Blood Glucose Concentrations: Population study in 1,013 eastern Finnish men. Diabetes Care 1997, 20, 426-428, 10.2337/diacare.20.3.426.
- Jukka T. Salonen; Tomi-Pekka Tuomainen; Kristiina Nyyssönen; Hanna-Maaria Lakka; Kari Punnonen; Relation between iron stores and non-insulin dependent diabetes in men: case-control study. BMJ 1998, 317, 727-730, 10.1136/bmj.317.7160.727.
- E S Ford; M. E. Cogswell; Diabetes and serum ferritin concentration among U.S. adults.. Diabetes Care 1999, 22, 1978-1983, 10.2337/diacare.22.12.1978.
- Forouhi, N.G.; Harding, A.H.; Allison, M.; Sandhu, M.S.; Welch, A.; Luben, R.; Bingham, S.; Khaw, K.T.; Wareham, N.J.; Elevated serum ferritin levels predict new-onset type 2 diabetes: Results from the EPIC-Norfolk prospective study. . Diabetologia 2007, 50, 949–956, 10.1007/s00125-007-0604-5.
- Luca Valenti; Anna Ludovica Fracanzani; Elisabetta Bugianesi; Paola Dongiovanni; Enrico Galmozzi; Ester Vanni; Elena Canavesi; Ezio Lattuada; Giancarlo Roviaro; Giulio Marchesini; et al.Silvia Fargion HFE Genotype, Parenchymal Iron Accumulation, and Liver Fibrosis in Patients With Nonalcoholic Fatty Liver Disease. Gastroenterology 2010, 138, 905-912, 10.1053/j.gastro.2009.11.013.
- Miguel Arredondo; Marcela Fuentes; Denisse Jorquera; Valeria Candia; Elena Carrasco; Elba Leiva; Veronica Mujica; Eva Hertrampf; Francisco Pérez; Cross-Talk Between Body Iron Stores and Diabetes: Iron Stores are Associated with Activity and Microsatellite Polymorphism of the Heme Oxygenase and Type 2 Diabetes. Biological Trace Element Research 2010, 143, 625-636, 10.1007/s12011-010-8895-7.
- Raffaela Rametta; Anna Ludovica Fracanzani; Silvia Fargion; Paola Dongiovanni; Dysmetabolic Hyperferritinemia and Dysmetabolic Iron Overload Syndrome (DIOS): Two Related Conditions or Different Entities?. Current Pharmaceutical Design 2020, 26, 1025-1035, 10.2174/1381612826666200131103018.
- C. Niederau; M. Berger; W. Stremmel; A. Starke; G. Strohmeyer; R. Ebert; E. Siegel; W. Creutzfeldt; Hyperinsulinaemia in non-cirrhotic haemochromatosis: impaired hepatic insulin degradation?. Diabetologia 1984, 26, 441-444, 10.1007/bf00262217.
- Silvia Fargion; P. Dongiovanni; A. Guzzo; S. Colombo; L. Valenti; A. L. Fracanzani; Iron and insulin resistance. Alimentary Pharmacology and Therapeutics 2005, 22, 61-63, 10.1111/j.1365-2036.2005.02599.x.
- Luca Valenti; Anna Ludovica Fracanzani; Paola Dongiovanni; Elisabetta Bugianesi; Giulio Marchesini; Paola Manzini; Ester Vanni; Silvia Fargion; Iron Depletion by Phlebotomy Improves Insulin Resistance in Patients With Nonalcoholic Fatty Liver Disease and Hyperferritinemia: Evidence from a Case-Control Study. The American Journal of Gastroenterology 2007, 102, 1251-1258, 10.1111/j.1572-0241.2007.01192.x.
- Facchini, F.S.; Effect of phlebotomy on plasma glucose and insulin concentrations. . Diabetes Care 1998,, 21, 2190.
- Luca Valenti; Paola Dongiovanni; Anna Ludovica Fracanzani; Silvia Fargion; Bloodletting ameliorates insulin sensitivity and secretion in parallel to reducing liver iron in carriers of HFE gene mutations: response to Equitani et al.. Diabetes Care 2008, 31, e18-e18, 10.2337/dc07-2181.
- Francesco Equitani; José-Manuel Fernández-Real; Giacomo Menichella; Maurizio Koch; Menotti Calvani; Valerio Nobili; Geltrude Mingrone; Melania Manco; Bloodletting Ameliorates Insulin Sensitivity and Secretion in Parallel to Reducing Liver Iron in Carriers of HFE Gene Mutations. Diabetes Care 2007, 31, 3-8, 10.2337/dc07-0939.
- 76. Dongiovanni, P.; Valenti, L.; Ludovica Fracanzani, A.; Gatti, S.; Cairo, G.; Fargion, S.; Iron depletion by deferoxamine up-regulates glucose uptake and insulin signaling in hepatoma cells and in rat liver. . Am. J. Pathol. 2008, 172, 738–747, 10.2353/ajpath.2008.070097.
- G. L. Semenza; Regulation of Metabolism by Hypoxia-Inducible Factor 1. Cold Spring Harbor Symposia on Quantitative Biology 2011, 76, 347-353, 10.1101/sqb.2011.76.010678.
- Chiara Macchi; Liliana Steffani; Roberto Oleari; Antonella Lettieri; Luca Valenti; Paola Dongiovanni; A Romero-Ruiz; Manuel Tena-Sempere; Anna Cariboni; Paolo Magni; et al.Massimiliano Ruscica Iron overload induces hypogonadism in male mice via extrahypothalamic mechanisms. Molecular and Cellular Endocrinology 2017, 454, 135-145, 10.1016/j.mce.2017.06.019.
- Antonello Pietrangelo; Hemochromatosis: An endocrine liver disease. Hepatology 2007, 46, 1291-1301, 10.1002/hep.21886.
- Dongiovanni, P.; Fracanzani, A.L.; Fargion, S.; Valenti, L.; . Iron in fatty liver and in the metabolic syndrome: A promising therapeutic target.. J. Hepatol. 2011, 55, 920–932, 10.1016/j.jhep.2011.05.008.
- Raffaela Rametta; Paola Dongiovanni; Serena Pelusi; Paolo Francione; Federica Iuculano; Vittorio Borroni; Erika Fatta; Annalisa Castagna; Domenico Girelli; Silvia Fargion; et al.Luca Valenti Hepcidin resistance in dysmetabolic iron overload. Liver International 2016, 36, 1540-1548, 10.1111/liv.13124.
- David D. Kitts; Bioactive substances in food: identification and potential uses. Canadian Journal of Physiology and Pharmacology 1994, 72, 423-434, 10.1139/y94-062.
- Hurrell, R.; Egli, I.; Iron bioavailability and dietary reference values. . Am. J. Clin. Nutr. 2010, 91, 1461S–1467S, 10.3945/ajcn.2010.28674F.
- Christina N. Kontoghiorghe; Annita Kolnagou; George J. Kontoghiorghes; Phytochelators Intended for Clinical Use in Iron Overload, Other Diseases of Iron Imbalance and Free Radical Pathology. Molecules 2015, 20, 20841-20872, 10.3390/molecules201119725.
- Anwesha Bhowmik; Durbadal Ojha; Debayan Goswami; Rashmi Das; Nidhi S. Chandra; Tapan K. Chatterjee; Amit Chakravarty; Sudipa Chakravarty; Debprasad Chattopadhyay; Inositol hexa phosphoric acid (phytic acid), a nutraceuticals, attenuates iron-induced oxidative stress and alleviates liver injury in iron overloaded mice. Biomedicine & Pharmacotherapy 2017, 87, 443-450, 10.1016/j.biopha.2016.12.125.
- Ulrich Schlemmer; Wenche Frølich; Rafel M. Prieto; Félix Grases; Phytate in foods and significance for humans: Food sources, intake, processing, bioavailability, protective role and analysis. Molecular Nutrition & Food Research 2009, 53, S330-S375, 10.1002/mnfr.200900099.
- 86. Cosgrove, D.J. . Inositol Phosphates; Elsevier: Amsterdam, The Netherlands, 1980; pp. 1.
- Mats Brune; Lena Rossander-Hultén; Leif Hallberg; Ann Gleerup; Ann-Sofie Sandberg; Iron Absorption from Bread in Humans: Inhibiting Effects of Cereal Fiber, Phytate and Inositol Phosphates with Different Numbers of Phosphate Groups. The Journal of Nutrition 1992, 122, 442-449, 10.1093/jn/122.3.442.
- L Hallberg; M Brune; L Rossander; Iron absorption in man: ascorbic acid and dose-dependent inhibition by phytate. The American Journal of Clinical Nutrition 1989, 49, 140-144, 10.1093/ajcn/49.1.140.
- R F Hurrell; M A Juillerat; M B Reddy; S R Lynch; S A Dassenko; J D Cook; Soy protein, phytate, and iron absorption in humans. The American Journal of Clinical Nutrition 1992, 56, 573-578, 10.1093/ajcn/56.3.573.
- Véronique Cheynier; Gilles Comte; Kevin M. Davies; Vincenzo Lattanzio; Stefan Martens; Plant phenolics: Recent advances on their biosynthesis, genetics, and ecophysiology. Plant Physiology and Biochemistry 2013, 72, 1-20, 10.1016/j.plaphy.2013.05.009.
- Brune, M.; Rossander, L.; Hallberg, L.; Iron absorption and phenolic compounds: Importance of different phenolic structures. . Eur. J. Clin. Nutr. 1989, 43, 547–557.
- Watson, R.R.; Preedy, V.R.; Zibadi, S. . Polyphenols in Human Health and Disease; Academic Press: Cambridge, MA, USA, 2014.: Cambridge, MA, USA, 2014; pp. 1.
- Tuntawiroon, M.; Sritongkul, N.; Brune, M.; Rossander-Hulten, L.; Pleehachinda, R.; Suwanik, R.; Hallberg, L.; Dose-dependent inhibitory effect of phenolic compounds in foods on nonheme-iron absorption in men. . Am. J. Clin. Nutr. 1991, 53, 554–557, 10.1093/ajcn/53.2.554..
- D J Fleming; P F Jacques; G E Dallal; Katherine L. Tucker; P W Wilson; R J Wood; Dietary determinants of iron stores in a free-living elderly population: The Framingham Heart Study.. The American Journal of Clinical Nutrition 1998, 67, 722-733, 10.1093/ajcn/67.4.722.
- Hurrell, R.F.; Reddy, M.; Cook, J.D.; . Inhibition of non-haem iron absorption in man by polyphenolic-containing beverages.. Br. J. Nutr. 1999, 81, 289–295.
- P B Disler; S R Lynch; R W Charlton; J D Torrance; T H Bothwell; R B Walker; F Mayet; The effect of tea on iron absorption.. Gut 1975, 16, 193-200, 10.1136/gut.16.3.193.
- L Rossander; L Hallberg; E Björn-Rasmussen; Absorption of iron from breakfast meals. The American Journal of Clinical Nutrition 1979, 32, 2484-2489, 10.1093/ajcn/32.12.2484.
- Kaltwasser, J.P.; Werner, E.; Schalk, K.; Hansen, C.; Gottschalk, R.; Seidl, C.; Clinical trial on the effect of regular tea drinking on iron accumulation in genetic haemochromatosis.. Gut 1998, 43, 699-704, 10.1136/gut.43.5.699.
- S. Samman; BrittMarie Sandström; Maja Bjørndal Toft; K. Bukhave; Mikael Jensen; Sven S Sørensen; Marianne Hansen; Green tea or rosemary extract added to foods reduces nonheme-iron absorption.. The American Journal of Clinical Nutrition 2001, 73, 607-612, 10.1093/ajcn/73.3.607.
- Marija Lesjak; Rukshana Hoque; Sara Balesaria; Vernon Skinner; Edward S. Debnam; Surjit K. S. Srai; Paul A. Sharp; Quercetin Inhibits Intestinal Iron Absorption and Ferroportin Transporter Expression In Vivo and In Vitro. PLOS ONE 2014, 9, e102900, 10.1371/journal.pone.0102900.
- Marija Lesjak; Sara Balesaria; Vernon Skinner; Edward S. Debnam; Surjit Kaila S. Srai; Quercetin inhibits intestinal non-haem iron absorption by regulating iron metabolism genes in the tissues.. European Journal of Nutrition 2018, 58, 743-753, 10.1007/s00394-018-1680-7.
- Zohreh Sajadi Hezaveh; Azita Azarkeivan; Leila Janani; Sharieh Hosseini; Farzad Shidfar; The effect of quercetin on iron overload and inflammation in β-thalassemia major patients: A double-blind randomized clinical trial.. Complementary Therapies in Medicine 2019, 46, 24-28, 10.1016/j.ctim.2019.02.017.
- Hutchinson, C.; Bomford, A.; Geissler, C.A.; The iron-chelating potential of silybin in patients with hereditary haemochromatosis.. Eur. J. Clin. Nutr. 2010, 64, 1239–1241, 10.1038/ejcn.2010.136.
- Wu, T.-H.; Grape Seed Proanthocyanidin Extract Chelates Iron and Attenuates the Toxic Effects of 6-Hydroxydopamine: Implications for Parkinson’s Disease. . J. Food Biochem. 2010 2010, 34, 1, 10.1111/j.1745-4514.2009.00276.x..
- Hervé Lobbes; Cécile Gladine; Andrzej Mazur; Bruno Pereira; Christian Dualé; Jean-Michel Cardot; Marc Ruivard; Effect of procyanidin on dietary iron absorption in hereditary hemochromatosis and in dysmetabolic iron overload syndrome: A crossover double-blind randomized controlled trial. Clinical Nutrition 2020, 39, 97-103, 10.1016/j.clnu.2019.02.012.
- J D Cook; S A Dassenko; P Whittaker; Calcium supplementation: effect on iron absorption. The American Journal of Clinical Nutrition 1991, 53, 106-111, 10.1093/ajcn/53.1.106.
- Hallberg, L.; Rossander-Hulten, L.; Brune, M.; Gleerup, A.; . Calcium and iron absorption: Mechanism of action and nutritional importance. . Eur. J. Clin. Nutr. 1992, 46, 317–327.
- Zamzam K (Fariba) Roughead; Carol A Zito; J R Hunt; Inhibitory effects of dietary calcium on the initial uptake and subsequent retention of heme and nonheme iron in humans: comparisons using an intestinal lavage method. The American Journal of Clinical Nutrition 2005, 82, 589-597, 10.1093/ajcn.82.3.589.
- Barton, J.C.; Conrad, M.E.; Parmley, R.T.; Calcium inhibition of inorganic iron absorption in rats. . Gastroenterology 1983, 84, 90–101.
- Ben Thompson; Paul A. Sharp; Ruan Elliott; Susan Fairweather-Tait; Inhibitory Effect of Calcium on Non-heme Iron Absorption May Be Related to Translocation of DMT-1 at the Apical Membrane of Enterocytes. Journal of Agricultural and Food Chemistry 2010, 58, 8414-8417, 10.1021/jf101388z.
- B Dawson-Hughes; F H Seligson; V A Hughes; Effects of calcium carbonate and hydroxyapatite on zinc and iron retention in postmenopausal women. The American Journal of Clinical Nutrition 1986, 44, 83-88, 10.1093/ajcn/44.1.83.
- Sean R Lynch; The effect of calcium on iron absorption. Nutrition Research Reviews 2000, 13, 141-158, 10.1079/095442200108729043.
- S. M. Robinson; Keith M. Godfrey; Jonathan Denne; Vanessa Cox; The determinants of iron status in early pregnancy.. British Journal of Nutrition 1998, 79, 249-255, 10.1079/bjn19980042.
- J D Cook; E R Monsen; Food iron absorption in human subjects. III. Comparison of the effect of animal proteins on nonheme iron absorption. The American Journal of Clinical Nutrition 1976, 29, 859-867, 10.1093/ajcn/29.8.859.
- R F Hurrell; S R Lynch; T P Trinidad; S A Dassenko; J D Cook; Iron absorption in humans as influenced by bovine milk proteins. The American Journal of Clinical Nutrition 1989, 49, 546-552, 10.1093/ajcn/49.3.546.
- R F Hurrell; S R Lynch; T P Trinidad; S A Dassenko; J D Cook; Iron absorption in humans: bovine serum albumin compared with beef muscle and egg white. The American Journal of Clinical Nutrition 1988, 47, 102-107, 10.1093/ajcn/47.1.102.
- Palle Pedersen; Nils Milman; Extrinsic factors modifying expressivity of the HFE variant C282Y, H63D, S65C phenotypes in 1,294 Danish men. Blut Zeitschrift für die gesamte Blutforschung 2009, 88, 957-965, 10.1007/s00277-009-0714-x.
- S R Lynch; S A Dassenko; J D Cook; M A Juillerat; R F Hurrell; Inhibitory effect of a soybean-protein–related moiety on iron absorption in humans. The American Journal of Clinical Nutrition 1994, 60, 567-572, 10.1093/ajcn/60.4.567.
- Marı́a Nieves Garcı́a-Casal; Miguel Layrisse; Liseti Solano; Marı́a Adela Barón; Franklin Arguello; Daisy Llovera; José Ramı́rez; Irene Leets; Eleonora Tropper; María Nieves García-Casal; et al.María Adela BarónJosé Ramírez Vitamin A and β-Carotene Can Improve Nonheme Iron Absorption from Rice, Wheat and Corn by Humans. The Journal of Nutrition 1998, 128, 646-650, 10.1093/jn/128.3.646.
- M W Bloem; M Wedel; E J Van Agtmaal; A J Speek; S Saowakontha; W H Schreurs; Vitamin A intervention: short-term effects of a single, oral, massive dose on iron metabolism. The American Journal of Clinical Nutrition 1990, 51, 76-79, 10.1093/ajcn/51.1.76.
- R E Hodges; H E Sauberlich; J E Canham; D L Wallace; R B Rucker; L A Mejía; M Mohanram; Hematopoietic studies in vitamin A deficiency. The American Journal of Clinical Nutrition 1978, 31, 876-885, 10.1093/ajcn/31.5.876.
- Dreyfuss, M.L.; Stoltzfus, R.J.; Shrestha, J.B.; Pradhan, E.K.; LeClerq, S.C.; Khatry, S.K.; Shrestha, S.R.; Katz, J.; Albonico, M.; West, K.P., Jr.; et al. Hookworms, malaria and vitamin A deficiency contribute to anemia and iron deficiency among pregnant women in the plains of Nepal. . J. Nutr. 2000, 130, 2527–2536, 10.1093/jn/130.10.2527..
- P. Brissot; A. Le Treut; G. Dien; M. Cottencin; M. Simon; M. Bourel; Hypovitaminemia A in Idiopathic Hemochromatosis and Hepatic Cirrhosis. Digestion 1978, 17, 469-478, 10.1159/000198153.
- G.D. Kom; E. Schwedhelm; P. Nielsen; R.H. Böger; Increased urinary excretion of 8-iso-prostaglandin F2α in patients with HFE-related hemochromatosis: A case-control study. Free Radical Biology and Medicine 2006, 40, 1194-1200, 10.1016/j.freeradbiomed.2005.11.004.
- Gardner, R.; Hodges, R.; Rucker, R. . Fate of erythrocyte iron in vitamin A deficient rats. In Federation Proceedings; FASEB: Bethesda, MD, USA, 1979; pp. Volume 38.
- L A Mejía; R E Hodges; R B Rucker; Clinical signs of anemia in vitamin A-deficient rats. The American Journal of Clinical Nutrition 1979, 32, 1439-1444, 10.1093/ajcn/32.7.1439.
- K. W. Sijtsma; G. J. Van Den Berg; A. G. Lemmens; C. E. West; A. C. Beynen; Iron status in rats fed on diets containing marginal amounts of vitamin A. British Journal of Nutrition 1993, 70, 777-785, 10.1079/bjn19930173.
- Annet J. C. Roodenburg; Clive E. West; Shiguang Yu; Anton C. Beynen; Comparison between time-dependent changes in iron metabolism of rats as induced by marginal deficiency of either vitamin A or iron. British Journal of Nutrition 1994, 71, 687-699, 10.1079/bjn19940176.
- Marta Citelli; Luciana Linhares Bittencourt; Simone Vargas Da Silva; Anna Paola Trindade Rocha Pierucci; Cristiana Pedrosa; Vitamin A Modulates the Expression of Genes Involved in Iron Bioavailability. Biological Trace Element Research 2012, 149, 64-70, 10.1007/s12011-012-9397-6.
- Oksana Katz; Ram Reifen; Aaron Lerner; β-Carotene can reverse dysregulation of iron protein in an in vitro model of inflammation. Immunologic Research 2014, 61, 70-78, 10.1007/s12026-014-8570-8.
- Sean R. Lynch; James D. Cook; INTERACTION OF VITAMIN C AND IRON. Annals of the New York Academy of Sciences 1980, 355, 32-44, 10.1111/j.1749-6632.1980.tb21325.x.
- Krishna Pillay Madhavan Nair; Ginnela N.V. Brahmam; Madhari S. Radhika; Roy Choudhury Dripta; Punjal Ravinder; Nagalla Balakrishna; Zhensheng Chen; Keli Hawthorne; Steven A. Abrams; Inclusion of guava enhances non-heme iron bioavailability but not fractional zinc absorption from a rice-based meal in adolescents.. The Journal of Nutrition 2013, 143, 852-8, 10.3945/jn.112.171702.
- Hallberg, L.; Rossander, L.; Effect of different drinks on the absorption of non-heme iron from composite meals. . Hum. Nutr. Appl. Nutr. 1982, 36, 116–123.
- Hallberg, L.; Brune, M.; Rossander, L.; Effect of ascorbic acid on iron absorption from different types of meals. Studies with ascorbic-acid-rich foods and synthetic ascorbic acid given in different amounts with different meals.. Hum. Nutr. Appl. Nutr. 1986, 40, 97–113.
- Margarita Diaz; Jorge L Rosado; Lindsay H Allen; Steve Abrams; Olga P García; The efficacy of a local ascorbic acid–rich food in improving iron absorption from Mexican diets: a field study using stable isotopes. The American Journal of Clinical Nutrition 2003, 78, 436-440, 10.1093/ajcn/78.3.436.
- Prashanth Thankachan; Thomas Walczyk; Sumithra Muthayya; Anura V Kurpad; Richard F Hurrell; Iron absorption in young Indian women: the interaction of iron status with the influence of tea and ascorbic acid. The American Journal of Clinical Nutrition 2008, 87, 881-886, 10.1093/ajcn/87.4.881.
- J D Cook; E R Monsen; Vitamin C, the common cold, and iron absorption. The American Journal of Clinical Nutrition 1977, 30, 235-241, 10.1093/ajcn/30.2.235.
- D Siegenberg; R D Baynes; T H Bothwell; B J Macfarlane; R D Lamparelli; N G Car; P MacPhail; U Schmidt; A Tal; F Mayet; et al. Ascorbic acid prevents the dose-dependent inhibitory effects of polyphenols and phytates on nonheme-iron absorption. The American Journal of Clinical Nutrition 1991, 53, 537-541, 10.1093/ajcn/53.2.537.
- A Stekel; M Olivares; F Pizarro; P Chadud; I Lopez; M Amar; Absorption of fortification iron from milk formulas in infants. The American Journal of Clinical Nutrition 1986, 43, 917-922, 10.1093/ajcn/43.6.917.
- Ballot, D.; Baynes, R.D.; Bothwell, T.H.; Gillooly, M.; MacFarlane, B.J.; MacPhail, A.P.; Lyons, G.; Derman, D.P.; Bezwoda, W.R.; Torrance, J.D.; et al.et al. The effects of fruit juices and fruits on the absorption of iron from a rice meal. . Br. J. Nutr. 1987, 57, 331–343, 10.1079/bjn19870041.
- M. Gillooly; T. H. Bothwell; J. D. Torrance; A. P. MacPhail; D. P. Derman; W. R. Bezwoda; W. Mills; R. W. Charlton; Fatima Mayet; The effects of organic acids, phytates and polyphenols on the absorption of iron from vegetables. British Journal of Nutrition 1983, 49, 331-342, 10.1079/bjn19830042.
- Seshadri, S.; Shah, A.; Bhade, S.; Haematologic response of anaemic preschool children to ascorbic acid supplementation.. Hum. Nutr. Appl. Nutr. 1985, 39, 151–154.
- Mao, X.; Yao, G.; Effect of vitamin C supplementations on iron deficiency anemia in Chinese children.. Biomed. Environ. Sci. Bes 1992, 5, 125–129.
- Kathryn L. Beck; Cathryn A. Conlon; Rozanne Kruger; Jane Coad; Welma Stonehouse; Gold kiwifruit consumed with an iron-fortified breakfast cereal meal improves iron status in women with low iron stores: a 16-week randomised controlled trial. British Journal of Nutrition 2010, 105, 101-109, 10.1017/s0007114510003144.
- J R Hunt; S K Gallagher; L K Johnson; Effect of ascorbic acid on apparent iron absorption by women with low iron stores. The American Journal of Clinical Nutrition 1994, 59, 1381-1385, 10.1093/ajcn/59.6.1381.
- Olga P García; Margarita Diaz; Jorge L Rosado; L H Allen; Ascorbic acid from lime juice does not improve the iron status of iron-deficient women in rural Mexico. The American Journal of Clinical Nutrition 2003, 78, 267-273, 10.1093/ajcn/78.2.267.
- L Hallberg; L Rossander; Improvement of iron nutrition in developing countries: comparison of adding meat, soy protein, ascorbic acid, citric acid, and ferrous sulphate on iron absorption from a simple Latin American-type of meal. The American Journal of Clinical Nutrition 1984, 39, 577-583, 10.1093/ajcn/39.4.577.
- Jacklyn Jackson; Rebecca Haslam; Mark McEvoy; Lesley MacDonald-Wicks; Amanda J. Patterson; Is Higher Consumption of Animal Flesh Foods Associated with Better Iron Status among Adults in Developed Countries? A Systematic Review. Nutrients 2016, 8, 89, 10.3390/nu8020089.
- Miguel Layrisse; Carlos Martínez-Torres; Marcel Roche; Effect of Interaction of Various Foods on Iron Absorption. The American Journal of Clinical Nutrition 1968, 21, 1175-1183, 10.1093/ajcn/21.10.1175.
- Sean R. Lynch; Richard F. Hurrell; Sandra A. Dassenko; James D. Cook; The Effect of Dietary Proteins on Iron Bioavailability in Man. Plant Promoters and Transcription Factors 1989, 249, 117-132, 10.1007/978-1-4684-9111-1_8.
- Bjorn-Rasmussen, E.; Hallberg, L.; Effect of animal proteins on the absorption of food iron in man. . Nutr. Metab. 1979, 23, 192–202, 10.1159/000176256.
- Zhang, D.; Carpenter, C.E.; Mahoney, A.W.; A mechanistic hypothesis for meat enhancement of nonheme iron absorption: Stimulation of gastric secretions and iron chelation. . Nutr. Res. 1990, 10, 929–935.
- Richard F. Hurrell; Manju B. Reddy; Marcel Juillerat; James D. Cook; Meat Protein Fractions Enhance Nonheme Iron Absorption in Humans. The Journal of Nutrition 2006, 136, 2808-2812, 10.1093/jn/136.11.2808.
- S Storcksdieck Genannt Bonsmann; Genannt Bonsmann; R. F. Hurrell; Stefan Storcksdieck Genannt Bonsmann; Iron-Binding Properties, Amino Acid Composition, and Structure of Muscle Tissue Peptides from in vitro Digestion of Different Meat Sources. Journal of Food Science 2007, 72, S019-S029, 10.1111/j.1750-3841.2006.00229.x.
- Charlotte N. Armah; Paul Sharp; Fred A. Mellon; Sandra Pariagh; Elizabeth K. Lund; J R Dainty; Birgit Teucher; Susan Fairweather-Tait; L-α-Glycerophosphocholine Contributes to Meat's Enhancement of Nonheme Iron Absorption. The Journal of Nutrition 2008, 138, 873-877, 10.1093/jn/138.5.873.
- Etsuo Niki; Maret G. Traber; A History of Vitamin E. Annals of Nutrition and Metabolism 2012, 61, 207-212, 10.1159/000343106.
- Fatemeh Khadangi; Angelo Azzi; Vitamin E – The Next 100 Years. IUBMB Life 2019, 71, 411-415, 10.1002/iub.1990.
- Lisa Schmölz; Marc Birringer; Stefan Lorkowski; Maria Wallert; Complexity of vitamin E metabolism. World Journal of Biological Chemistry 2016, 7, 14-43, 10.4331/wjbc.v7.i1.14.
- Bruce R. Bacon; Robert S. Britton; The pathology of hepatic iron overload: A free radical-Mediated Process?. Hepatology 1990, 11, 127-137, 10.1002/hep.1840110122.
- Bruce R. Bacon; John F. Healey; Gary M. Brittenham; C.H. Park; Jodi Nunnari; Anthony S. Tavill; Herbert L. Bonkovsky; Hepatic microsomal function in rats with chronic dietary iron overload. Gastroenterology 1986, 90, 1844-1853, 10.1016/0016-5085(86)90251-9.
- Bruce R. Bacon; Chanho H. Park; Gary M. Brittenham; Rosemary O-Neill; Anthony S. Tavill; Hepatic mitochondrial oxidative metabolism in rats with chronic dietary iron overload. Hepatology 1985, 5, 789-797, 10.1002/hep.1840050514.
- B M Myers; F G Prendergast; R Holman; S M Kuntz; N F LaRusso; Alterations in the structure, physicochemical properties, and pH of hepatocyte lysosomes in experimental iron overload.. Journal of Clinical Investigation 1991, 88, 1207-1215, 10.1172/jci115423.
- J. M. C. Gutteridge; D. A. Rowley; E. Griffiths; B. Halliwell; Low-molecular-weight iron complexes and oxygen radical reactions in idiopathic haemochromatosis. Clinical Science 1985, 68, 463-467, 10.1042/cs0680463.
- E Cadenas; M Ginsberg; U Rabe; H Sies; Evaluation of α-tocopherol antioxidant activity in microsomal lipid peroxidation as detected by low-level chemiluminescence. Biochemical Journal 1984, 223, 755-759, 10.1042/bj2230755.
- Graham W. Burton; Anne Joyce; Keith U. Ingold; Is vitamin E the only lipid-soluble, chain-breaking antioxidant in human blood plasma and erythrocyte membranes?. Archives of Biochemistry and Biophysics 1983, 221, 281-290, 10.1016/0003-9861(83)90145-5.
- Helmut Sies; Michael E. Murphy; Role of tocopherols in the protection of biological systems against oxidative damage. Journal of Photochemistry and Photobiology B: Biology 1991, 8, 211, 10.1016/1011-1344(91)80061-l.
- B. Halliwell; JohnM.C. Gutteridge; LIPID PEROXIDATION, OXYGEN RADICALS, CELL DAMAGE, AND ANTIOXIDANT THERAPY. The Lancet 1984, 323, 1396-1397, 10.1016/s0140-6736(84)91886-5.
- A J Dabbagh; T Mannion; S M Lynch; B Frei; The effect of iron overload on rat plasma and liver oxidant status in vivo. Biochemical Journal 1994, 300, 799-803, 10.1042/bj3000799.
- Ian S Young; Tom G. Trouton; Jonathan J. Torney; Dorothy McMaster; Michael E. Callender; Elisabeth R. Trimble; Antioxidant status and lipid peroxidation in hereditary haemochromatosis. Free Radical Biology and Medicine 1994, 16, 393-397, 10.1016/0891-5849(94)90041-8.
- von Herbay, A.; de Groot, H.; Hegi, U.; Stremmel, W.; Strohmeyer, G.; Sies, H.; Low vitamin E content in plasma of patients with alcoholic liver disease, hemochromatosis and Wilson's disease.. J. Hepatol. 1994, 20, 41–46.
- K. E. Brown; J. E. Poulos; L. Li; A. M. Soweid; G. A. Ramm; R. O'neill; R. S. Britton; B. R. Bacon; Effect of vitamin E supplementation on hepatic fibrogenesis in chronic dietary iron overload. American Journal of Physiology-Gastrointestinal and Liver Physiology 1997, 272, G116-G123, 10.1152/ajpgi.1997.272.1.g116.
- Whittaker, P.; Wamer, W.G.; Chanderbhan, R.F.; Dunkel, V.C.; Effects of alpha-tocopherol and beta-carotene on hepatic lipid peroxidation and blood lipids in rats with dietary iron overload. . Nutr. Cancer. 1996, 25, 119–128, 10.1080/01635589609514434.
- Nadia Ferlazzo; Giuseppa Visalli; Santa Cirmi; Giovanni Enrico Lombardo; Pasqualina Laganà; Angela Di Pietro; Michele Navarra; Natural iron chelators: Protective role in A549 cells of flavonoids-rich extracts of Citrus juices in Fe 3+ -induced oxidative stress. Environmental Toxicology and Pharmacology 2016, 43, 248-256, 10.1016/j.etap.2016.03.005.
- Joo-Hyun Shin; Hyo-Jin Jeon; Jihye Park; Mi-Sook Chang; Epigallocatechin-3-gallate prevents oxidative stress-induced cellular senescence in human mesenchymal stem cells via Nrf2.. International Journal of Molecular Medicine 2016, 38, 1075-82, 10.3892/ijmm.2016.2694.
- Helmut Sies; Dean P. Jones; Reactive oxygen species (ROS) as pleiotropic physiological signalling agents.. Nature Reviews Molecular Cell Biology 2020, 21, 1-21, 10.1038/s41580-020-0230-3.
- Ma, Q.; Kim, E.Y.; Lindsay, E.A.; Han, O.; Bioactive dietary polyphenols inhibit heme iron absorption in a dose-dependent manner in human intestinal Caco-2 cells. . J. Food Sci. 2011, 76, H143–H150, 10.1111/j.1750-3841.2011.02184.x.
- Qiang Niu; Lati Mu; Shugang Li; Shangzhi Xu; Ruling Ma; Shuxia Guo; Proanthocyanidin Protects Human Embryo Hepatocytes from Fluoride-Induced Oxidative Stress by Regulating Iron Metabolism. Biological Trace Element Research 2015, 169, 174-179, 10.1007/s12011-015-0409-1.
- Jiao, Y.; Wilkinson, J.T.; Di, X.; Wang, W.; Hatcher, H.; Kock, N.D.; D’Agostino, R., Jr.; Knovich, M.A.; Torti, F.M.; Torti, S.V.; et al. Curcumin, a cancer chemopreventive and chemotherapeutic agent, is a biologically active iron chelator.. Blood 2009, 113, 462–469, 10.1182/blood-2008-05-155952.
- Naresh Kumar; Vikas Pruthi; Potential applications of ferulic acid from natural sources.. Biotechnology Reports 2014, 4, 86-93, 10.1016/j.btre.2014.09.002.
- Yang Qiao; Huan He; Zeyu Zhang; Zhangping Liao; Dong Yin; Dan Liu; Bo Yi; Ming He; Long-Term Sodium Ferulate Supplementation Scavenges Oxygen Radicals and Reverses Liver Damage Induced by Iron Overloading. Molecules 2016, 21, 1219, 10.3390/molecules21091219.
- Calamini, B.; Ratia, K.; Malkowski, M.G.; Cuendet, M.; Pezzuto, J.M.; Santarsiero, B.D.; Mesecar, A.D.; Pleiotropic mechanisms facilitated by resveratrol and its metabolites. . Biochem. J. 2010, 429, 273–282, 10.1042/BJ20091857.
- Subhash K. Das; Jessica Desaulniers; Jason R. B. Dyck; Zamaneh Kassiri; Gavin Y Oudit; Resveratrol mediates therapeutic hepatic effects in acquired and genetic murine models of iron-overload. Liver International 2015, 36, 246-257, 10.1111/liv.12893.
- Marica Meroni; Miriam Longo; Veronica Erconi; Luca Valenti; Stefano Gatti; Anna Ludovica Fracanzani; Paola Dongiovanni; mir-101-3p Downregulation Promotes Fibrogenesis by Facilitating Hepatic Stellate Cell Transdifferentiation During Insulin Resistance.. Nutrients 2019, 11, 2597, 10.3390/nu11112597.
- Bartel, D.P.; Genomics, biogenesis, mechanism, and function. . Cell 2004, 116, 281–297, 10.1016/s0092-8674(04)00045-5.
- Chiranjib Chakraborty; Ashish Ranjan Sharma; Garima Sharma; C. George Priya Doss; Sang-Soo Lee; Therapeutic miRNA and siRNA: Moving from Bench to Clinic as Next Generation Medicine. Molecular Therapy - Nucleic Acids 2017, 8, 132-143, 10.1016/j.omtn.2017.06.005.
- Paola Dongiovanni; Marica Meroni; Miriam Longo; Silvia Fargion; Anna Ludovica Fracanzani; miRNA Signature in NAFLD: A Turning Point for a Non-Invasive Diagnosis. International Journal of Molecular Sciences 2018, 19, 3966, 10.3390/ijms19123966.
- Céline Tiffon; The Impact of Nutrition and Environmental Epigenetics on Human Health and Disease. International Journal of Molecular Sciences 2018, 19, 3425, 10.3390/ijms19113425.
- Eric Huntzinger; Elisa Izaurralde; Gene silencing by microRNAs: contributions of translational repression and mRNA decay. Nature Reviews Microbiology 2011, 12, 99-110, 10.1038/nrg2936.
- Friedman, R.C.; Farh, K.K.; Burge, C.B.; Bartel, D.P.; Most mammalian mRNAs are conserved targets of microRNAs.. Genome Res. 2009, 19, 92–105, 10.1101/gr.082701.108.
- Mirco Castoldi; Martina U. Muckenthaler; Regulation of iron homeostasis by microRNAs. Cellular and Molecular Life Sciences 2012, 69, 3945-3952, 10.1007/s00018-012-1031-4.
- Michael Faller; Michio Matsunaga; Sheng Yin; Joseph A. Loo; Feng Guo; Heme is involved in microRNA processing. Nature Structural & Molecular Biology 2006, 14, 23-29, 10.1038/nsmb1182.
- Dale G. Schaar; Daniel J. Medina; Dirk F. Moore; Roger K. Strair; Yi Ting; miR-320 targets transferrin receptor 1 (CD71) and inhibits cell proliferation. Experimental Hematology 2009, 37, 245-255, 10.1016/j.exphem.2008.10.002.
- Yalin Liao; Xiaogu Du; Bo Lönnerdal; miR-214 Regulates Lactoferrin Expression and Pro-Apoptotic Function in Mammary Epithelial Cells. The Journal of Nutrition 2010, 140, 1552-1556, 10.3945/jn.110.124289.
- Liao, Y.; Lonnerdal, B.; miR-584 mediates post-transcriptional expression of lactoferrin receptor in Caco-2 cells and in mouse small intestine during the perinatal period.. Int. J. Biochem. Cell Biol. 2010, 42, 1363–1369, 10.1016/j.biocel.2009.07.019.
- Svitlana I. Shpyleva; Volodymyr Tryndyak; Olga Kovalchuk; Athena Starlard-Davenport; Vasyl’ F. Chekhun; Frederick A. Beland; Igor P. Pogribny; Role of ferritin alterations in human breast cancer cells. Breast Cancer Research and Treatment 2010, 126, 63-71, 10.1007/s10549-010-0849-4.
- Castoldi, M.; Vujic Spasic, M.; Altamura, S.; Elmen, J.; Lindow, M.; Kiss, J.; Stolte, J.; Sparla, R.; D'Alessandro, L.A.; Klingmuller, U.; et al.et al. The liver-specific microRNA miR-122 controls systemic iron homeostasis in mice.. J. Clin. Investig. 2011, 121, 1386–1396, 10.1172/jci44883.
- Masuko Kobori; Saeko Masumoto; Yukari Akimoto; Hideaki Oike; Chronic dietary intake of quercetin alleviates hepatic fat accumulation associated with consumption of a Western-style diet in C57/BL6J mice. Molecular Nutrition & Food Research 2010, 55, 530-540, 10.1002/mnfr.201000392.
- Takatoshi Murase; Koichi Misawa; Yoshihiko Minegishi; Masafumi Aoki; Hideo Ominami; Yasuto Suzuki; Yusuke Shibuya; Tadashi Hase; Coffee polyphenols suppress diet-induced body fat accumulation by downregulating SREBP-1c and related molecules in C57BL/6J mice. American Journal of Physiology-Endocrinology and Metabolism 2011, 300, E122-E133, 10.1152/ajpendo.00441.2010.

