 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Marcus Buschbeck | + 2609 word(s) | 2609 | 2021-04-12 07:54:40 | | | |
| 2 | Vivi Li | Meta information modification | 2609 | 2021-04-20 10:51:32 | | |
Video Upload Options
Mutations in genes encoding chromatin regulators are early events contributing to developing asymptomatic clonal hematopoiesis of indeterminate potential and its frequent progression to myeloid diseases with increasing severity. We focus on the subset of myeloid diseases encompassing myelodysplastic syndromes and their transformation to secondary acute myeloid leukemia. We introduce the major concepts of chromatin regulation that provide the basis of epigenetic regulation. In greater detail, we discuss those chromatin regulators that are frequently mutated in myelodysplastic syndromes. We discuss their role in the epigenetic regulation of normal hematopoiesis and the consequence of their mutation. Finally, we provide an update on the drugs interfering with chromatin regulation approved or in development for myelodysplastic syndromes and acute myeloid leukemia.
1. Introduction
Myelodysplastic syndromes (MDS) are part of a spectrum of clonal myeloid diseases starting with the asymptomatic expansion of mutated hematopoietic stem cell (HSC) clones and frequently ending with transformation to full-blown secondary acute myeloid leukemia (sAML) [1]. The evolution and progression of MDS and sAML is intimately linked to changes in the regulation of chromatin function and epigenetics. First, effector enzymes with epigenetic regulatory functions are among the most commonly mutated genes in MDS and AML [2][3]. Second, epigenetic abnormalities co-occur with genetic and cytogenetic changes in MDS and sAML, and together, contribute to the full manifestation of the disease [4]. Indeed, the accumulation of epigenetic changes has been suggested to represent a tipping point to transformation to sAML [1]. The fact that epigenetic changes are reversible has provided the rationale for developing therapies that target epigenetic regulators.
2. CHIP-MDS-sAML—A Spectrum Myeloid Diseases
The expansion of clonal populations of blood cells from a single hematopoietic stem cell (HSC) with one or more somatic mutations is divided into two categories age-related clonal hematopoiesis (ARCH) and clonal hematopoiesis of indeterminate potential (CHIP). ARCH describes broad recurrently occurring mutational events that can cause clonal hematopoiesis and lead to age-related pathologies, including inflammation, cancer mortality, as well as hematological malignancies [5]. On the other hand, CHIP is associated with detectable somatic clonal mutations in leukemia-driver genes with a variant allele frequency (VAF) of 2% or greater [6] (Figure 1). Individuals with CHIP show normal peripheral blood counts and no evidence of WHO-defined criteria for a hematological malignancy or other clonal disorders [7]. Mutations that also occur in MDS and sAML have been observed in healthy, mainly elderly populations as part of population-based studies [8][9]. CHIP-related mutational burden appears to increase with age, as CHIP is present in 10–15% of individuals aged over 70 years [1]. Interestingly, the most frequent mutations in CHIP affect the epigenetic regulators TET2, DNMT3A and ASXL1 and the splicing factor SF3B1. Individuals with CHIP have an increased risk of developing diseases of the lymphoid and myeloid lineage, including MDS. This happens when mutations increase the fitness of HSC clones allowing them to expand among the bulk HSC population, eventually resulting in clonal dominance. If mutations are coupled with reduced differentiation capacity, the expansion of mutated HSCs can lead to reduced generation of mature blood cells in one or several lineages (Figure 1). The current challenge lies in understanding how CHIP predisposes to developing disorders. For a more thorough discussion of CHIP and its consequences, please see recent reviews [5][6].
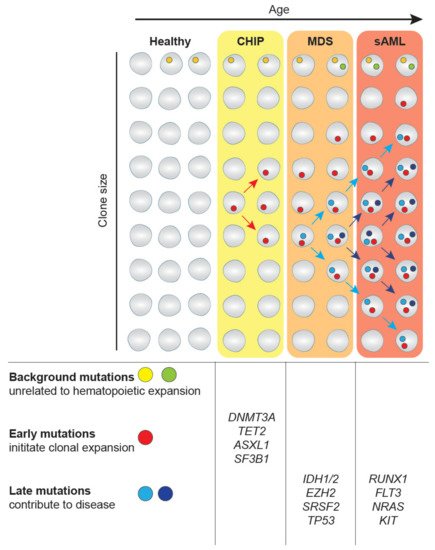
Figure 1. Clonal hematopoiesis in myelodysplastic syndromes (MDS) and transformation to secondary acute myeloid leukemia (sAML). Mutations in hematopoietic stem cell (HSC) clones occur at any time of our life as part of the aging process. While most mutations are background mutations that do not affect cellular properties, some mutations provide an advantage to HSCs, such as increased self-renewal. These mutations drive clonal expansion and the eventual development of the asymptomatic clonal hematopoiesis of indeterminate potential (CHIP). The further expansion frequently driven by the acquisition of additional genetic alterations can lead to MDS. The gain of additional driver mutations can further lead to transformation to sAML. This figure has been inspired by [10].
MDS is the most frequent hematopoietic disorder in the elderly [11][12]. Advanced age is the main contributing risk factor of acute myeloid malignancies, with the median age of diagnosis at around 70 years and 92% of MDS patients aged over 50 years [13][14]. MDS is characterized by the expansion of mutant HSC clones at the expense of normal hematopoiesis leading to low blast cell counts, but a substantial reduction of numbers of mature blood cell types referred to as cytopenias. Consequential symptoms are fatigue due to anemia [15], recurring infections related to neutrophil dysfunction [16] and autoimmune abnormalities, such as rheumatic heart disease [17].
Around 30% of MDS patients transform to sAML [18], which is characterized by further increases in blast cell counts above 20% in the bone marrow [19]. On the genetic and molecular level, sAML mutant HSC clones have acquired additional driver mutations that convert them into full leukemia stem cells (LSCs). These genetic alterations differ to some extent from other AML subtypes [20]. De novo AML occurs without any previous neoplasm, is more common in younger patients and is associated with better overall survival [21]. Compared to CHIP and early-stage MDS, LSCs in sAML and late-stage MDS have acquired mutations that confer uncontrolled growth, such as NRAS, and inhibition of apoptosis, such as TP53. Together with epigenetic abnormalities, these oncogenic mutations cause blast cell numbers to increase and inhibit differentiation, which is characteristic of the MDS-to-sAML transformation [1]. Furthermore, an abnormal stem cell niche in the bone marrow may favor the outgrowth of mutant clones and thus contribute to the disease [22][23].
In summary, MDS and sAML are part of a spectrum of clonal diseases affecting the myeloid lineage that can arise from CHIP. Mutations in epigenetic regulators are early events and provide a yet not fully understood function in disease etiology.
3. Epigenetic Regulators Frequently Mutated in Myeloid Diseases and Their Function
Recurrent mutations in CHIP, MDS and sAML affecting genes involved in epigenetic regulation include regulators of DNA methylation, histone modifiers and elements regulating higher-order chromatin architecture [2][24]. For these groups of genes, we discuss their normal role in hematopoiesis and the consequences of their mutations in the disease (summarized in Table 1). Again, we focus on MDS and sAML but also discuss selected insights from other types of AML.
Table 1. Mutations in epigenetic regulators in MDS and AML.
| Gene | Mutation Effect on Gene | Mutational Frequency | Characteristics |
|---|---|---|---|
| ASXL1 [25][26][27][28] | Loss-of-function mutation | 20% in MDS | Mutations enriched in elderly AML and sAML patients |
| 6–30% in AML | |||
| BCOR [29][30][31] | Loss-of-function mutation | 5% in MDS | Associated with poor prognosis |
| 9% in AML | |||
| DNMT3A [32][33][34][35][36][37] | Loss-of-function mutation | 13% in MDS | Thought to be initiating mutation during the pre-leukemic state |
| 20% in AML | Important for the balance of differentiation and self-renewal | ||
| EZH2 [38][39][40][41][42] | Loss-of-function mutation as well as gain of function mutations | 5% in MDS | Thought to regulate the balance between self-renewal and differentiation |
| 1–2% de novo AML | In MDS associated with poor prognosis | ||
| IDH1/2 [43][44][45][46][47][48] | Gain of function | 5% in MDS | Leads to the production of oncometabolite, which interferes with TET2 activity and histone demethylases |
| 20% in AML | IDH2 mutations are more common | ||
| RUNX1 [49][50][51][52][53][54] | Translocations | 10–20% in MDS | Significantly associated with EZH2 mutations |
| Loss-of-function mutation | 2–20% in AML | ||
| Cohesin [55][56][57][58][59][60] | Loss-of-function mutation | 10–15% in MDS, | Mutually exclusive |
| 10% in AML | often associated with mutations in NPM1, TET2, ASXL1 and EZH2 | ||
| TET2 [61][62][63][64][65][66][67][68] | Loss-of-function mutation | 30–50% in MDS | Important for myeloid differentiation and lineage commitment |
| 30% in sAML | Associated with poor prognosis in some studies |
3.1. Mutations Causing Aberrant DNA Methylation—TET2, DNMT3A, IDH
Advances in genome-wide DNA methylation studies have revealed distinct DNA methylation patterns at different stages of differentiation during hematopoiesis that demarcate myeloid and lymphoid lineage decisions [69][70]. In general, myelopoiesis is associated with a reduction of methylation marks. Genes methylated at their promoters in myeloid progenitor cells of mice were reported to become unmethylated in a lineage-specific manner. Examples are the neutrophil-specific gene, Mpo, encoding myeloperoxidase and Cxcr2 that encodes a chemokine to allow chemotaxis [71]. In contrast, lymphopoiesis depends on the maintenance of DNA methylation, as evidenced by a reduction in lymphoid progeny in mice with reduced Dnmt1 activity [71]. A principal characteristic of HSC is its life-long ability to self-renew. When DNMT1 activity is removed in mice, HSC and progenitors were reduced in the bone marrow, and differentiation patterns were disrupted, suggesting maintenance of DNA methylation plays a direct role in regulating HSC self-renewal and cell fate decisions [71]. Aberrant DNA methylation can often be seen in MDS and AML and is thought to drive disease progression [72]. In particular, mutations in TET2 and DNMT3A are frequently observed in the early stages of CHIP [7] and highlight the important role of aberrant DNA methylation, and not just hyper- or hypomethylation, in the contribution to myeloid malignancies [73].
DNMT3A establishes de novo DNA methylation, and it is thought that heterozygous mutant DNMT3A acts as a dominant-negative over wild-type DNMT3A, thereby reducing overall methyltransferase activity [32]. HSC of conditional Dnmt3a-knockout mice displays reduced differentiation capacities, while their self-renewal was elevated, which resulted in an accumulation of Dnmt3a-null HSCs in the bone marrow [33][34]. Similarly, in xenograft models, human DNMT3A-mutant HSCs demonstrated an advantage compared to wild-type HSCs, highlighting their contribution to a pre-leukemic state prior to the acquisition of additional mutations [35]. Indeed, DNMT3A mutations are one of the first ones to arise [36][37].
TET enzymes carry out antagonistic biochemical functions to DNMT3A [61]. TETs promote demethylation in an indirect manner involving oxidation of the methylated cytosine and base excision [62]. Deleterious TET2 mutations are common in hematologic malignancies, with 30–50% in patients with MDS and myeloproliferative neoplasia and 30% in sAML patients [63]. TET2 deficiency causes widespread hypermethylation in mice, where upregulated oncogenes and downregulated tumor suppressor genes may have contributed to the observed leukemogenesis [64]. Deletion of TET2 in CD34+CD38+ hematopoietic progenitor cells resulted in increased monocyte expansion, suggesting a role in myeloid differentiation or lineage commitment [65]. In various studies, the mutational status of TET2 has been associated with poor prognosis [66][67], while others could not demonstrate this association [63][68].
Isocitrate dehydrogenase (IDH) is a key enzyme in the citric acid cycle that catalyzes the conversion of isocitrate to 2-ketoglutarate, which is an important cofactor for TET enzymes and some histone demethylases [43]. IDH mutations are neomorphic mutations that change the enzymatic capacity resulting in the production of elevated levels of 2-hydroxyglutarate (2-HG), which acts as a competitive inhibitor of TETs and other 2-ketoglutarate-dependent enzymes, leading to a widespread increase in histone and DNA methylation [44][45]. IDH mutations block differentiation and promote LSCs to proliferate [46]. Mutations in IDH1 and IDH2 have been identified in around 5% of MDS cases [47], 9.7% of sAML and 20% of AML patients [43]. IDH1 mutations are less common than IDH2 mutations [47]. In IDH1, mutations can often be found on arginine R132 in the form of a cysteine (R132C) or histidine (R132H) substitution. In IDH2, the mutations affect arginine R140 or R172 replaced by glutamine (R140Q) or lysine (R172K), respectively. In myeloproliferative neoplasms and high-risk MDS, IDH mutations were linked to disease progression [48]. In contrast, in AML, the prognostic impact of IDH mutations could not be clearly determined and may depend on the specific point mutation and the presence or absence of co-mutations [43].
3.2. Dysregulation of Histone Modifications—EZH2, RUNX1, BCOR, ASXL1
The multimeric polycomb repressive complexes (PRC) 1 and 2 are histone writers that contribute to transcriptional silencing. PRC2 is responsible for all di- and tri-methylation of lysine 27 of H3 (H3K27me2/me3) that is mediated by its subunit EZH2 [74][75]. During lymphopoiesis, high expression levels of EHZ2 are associated with proliferating cells suggesting a role in lineage-specific cell cycle regulation [38]. H3K27me3 mediates the recruitment of PRC1 that mono-ubiquitylates H2A at lysine 119, inhibits transcriptional elongation and promotes chromatin compaction [76]. Interestingly, the PRC2-induced H3K27me3 mark is offset by the trithorax group (trxG), which mediates the activating H3K4me3 mark associated with open chromatin and gene activation [77]. Genes in loci that contain both marks are so-called “bivalent” domains that indicate flexible activation and repressive mechanisms. HSC contains many such bivalent genes [78]. Genome-wide changes of gene expression and histone modifications have shown HSC genes are “primed”’ for subsequent activation or repression during lineage commitment [79]. In this way, PRCs are thought to contribute to HSC self-renewal and maintenance of pluripotency by dynamically repressing cell fate regulators during hematopoiesis [39]. Mutations in EZH2, BCOR, ASXL1 and RUNX1 affect the function of PRCs.
Both loss and gain-of-function mutations of EZH2 are found in hematological disorders indicating a context-dependent function of EZH2 as an oncogene or tumor suppressor [39]. In MDS, primarily inactivating mutations of EZH2 occur in around 5% of patients [2] and are associated with poor prognosis [40] but not with progression to AML [80]. In de novo AML, loss-of-function mutations of EZH2 are less frequent and occur in 1–2% of patients [20]. Mechanistically, loss of Ezh2 in mice has been shown to promote MDS development by activating inflammatory cytokine responses resulting in impaired HSCs differentiation [41]. On the other hand, Ezh2-deficient mouse models have demonstrated the requirement of EZH2 for developing myeloid malignancies, including MLL-AF9 AML, in which Ezh2 mutation or deletion causes a loss of LSCs and an increase in differentiation [42].
While EZH2 is a component of PRC2, BCOR is a component of a variant of the PRC1 complex [29][81]. BCOR loss-of-function mutations occur in about 5% of cases of MDS and 9% of sAML patients and are associated with a poor prognosis [2][30]. Bcor loss results in myeloid progenitor expansion and the presence of oncogenic KrasG12D promotes leukemogenesis in mice [31].
ASXL1 forms a complex with BRCA1-associated protein 1 (BAP1) that physically interacts with PRC2 and deubiquitinylates histone H2A [25]. ASXL1 mutations lead to reduced levels of ASXL1 and are associated with a global reduction of PRC2 recruitment and H3K27me3 [25]. ASXL1 is mutated in approximately 20% of MDS patients, thus representing one of the top mutated genes [2]. In AML, ASXL1 mutations occur in 6–30% of patients and correlate with advancing age [26][27][28].
Mutations in ASXL1, EZH2 and BCOR1 are associated with mutations in the gene encoding the transcription factor RUNX1 [2]. With more than 50 reported translocations and various point mutations, RUNX1 is one of the most frequently mutated genes in AML [49][50]. In MDS, RUNX1 mutations occur in 10–20% of patients [51]. HSC self-renewal is disrupted in animals with mutated RUNX1 [52]. RUNX1 regulates the PU.1 gene, which is involved in developing all hematopoietic lineages. Disruption of normal RUNX1 activity results in PU.1 downregulation with various lineage-specific consequences, including an increased percentage of granulocytes in the bone marrow of mice [53]. While it is not fully clear how RUNX1 mutations synergize with mutations related to PRC function in disease, it is interesting to point out that RUNX1 protein can physically interact with PRCs and promote gene repression through their recruitment to gene promoters [54].
3.3. Altering Chromatin Structure—The Cohesin Complex
Somatic mutations affecting the cohesin complex have been identified in several diseases, including MDS and AML [82]. The cohesin complex consists of the core subunits SMC1, SMC3 and RAD21, which associate with either STAG1 or STAG2. One of its important functions is to align and stabilize sister chromatids during metaphase crucial for DNA replication, DNA repair and mitosis [55]. In addition, cohesin has an important role in the regulation of genome folding in interphase cells [56]. Loss-of-function cohesin mutations, mainly in the STAG2 gene, were detected in 10–15% of MDS and 20% of sAML patients and are associated with poor survival [57]. Interestingly, in several human leukemic cell lines, low expression of cohesin was observed, although no mutation could be identified [57]. On the mechanistic level, reduced cohesin function leads to changes in gene expression, possibly as a direct consequence of changes in chromatin architecture [58]. In particular, reduced sensitivity to inflammatory signals may affect the function of HSCs [59].
In conclusion, with mutations affecting cohesin, histone-modifying PRCs and the DNA methylation machinery, several central epigenetic mechanisms are perturbed in MDS and sAML. The common denominator of these mutations in disease is that they disrupt normal hematopoietic differentiation and promote the expansion of altered HSCs [57], thereby contributing to disease progression. The challenge for the field now is to identify specific vulnerabilities of mutant cells that can be exploited for therapeutic strategies aiming at synthetic lethality. An exciting example is a recent demonstration that cohesin mutant cells are hypersensitive to inhibitors of the DNA repair pathway [60].
References
- Sperling, A.S.; Gibson, C.J.; Ebert, B.L. The Genetics of Myelodysplastic Syndrome: From Clonal Haematopoiesis to Secondary Leukaemia. Nat. Rev. Cancer 2017, 17, 5–19.
- Haferlach, T.; Nagata, Y.; Grossmann, V.; Okuno, Y.; Bacher, U.; Nagae, G.; Schnittger, S.; Sanada, M.; Kon, A.; Alpermann, T.; et al. Landscape of Genetic Lesions in 944 Patients with Myelodysplastic Syndromes. Leukemia 2014, 28, 241–247.
- Shih, A.H.; Abdel-Wahab, O.; Patel, J.P.; Levine, R.L. The Role of Mutations in Epigenetic Regulators in Myeloid Malignancies. Nat. Rev. Cancer 2012, 12, 599–612.
- Issa, J.-P.J. The Myelodysplastic Syndrome as a Prototypical Epigenetic Disease. Blood 2013, 121.
- Shlush, L.I. Age-Related Clonal Hematopoiesis. Blood 2018, 131, 496–504.
- Steensma, D.P. Clinical Consequences of Clonal Hematopoiesis of Indeterminate Potential. Blood Adv. 2018, 2, 3404–3410.
- Steensma, D.P.; Bejar, R.; Jaiswal, S.; Lindsley, R.C.; Sekeres, M.A.; Hasserjian, R.P.; Ebert, B.L. Clonal Hematopoiesis of Indeterminate Potential and Its Distinction from Myelodysplastic Syndromes. Blood 2015, 126, 9–16.
- Genovese, G.; Kähler, A.K.; Handsaker, R.E.; Lindberg, J.; Rose, S.A.; Bakhoum, S.F.; Chambert, K.; Mick, E.; Neale, B.M.; Fromer, M.; et al. Clonal Hematopoiesis and Blood-Cancer Risk Inferred from Blood DNA Sequence. N. Engl. J. Med. 2014, 371, 2477–2487.
- Jaiswal, S.; Fontanillas, P.; Flannick, J.; Manning, A.; Grauman, P.V.; Mar, B.G.; Lindsley, R.C.; Mermel, C.H.; Burtt, N.; Chavez, A.; et al. Age-Related Clonal Hematopoiesis Associated with Adverse Outcomes. N. Engl. J. Med. 2014, 371, 2488–2498.
- Wouters, B.J.; Delwel, R. Epigenetics and Approaches to Targeted Epigenetic Therapy in Acute Myeloid Leukemia. Blood 2016, 127, 42–52.
- Kuendgen, A.; Strupp, C.; Aivado, M.; Hildebrandt, B.; Haas, R.; Gattermann, N.; Germing, U. Myelodysplastic Syndromes in Patients Younger Than Age 50. J. Clin. Oncol. 2006, 24, 5358–5365.
- Sekeres, M.A. Epidemiology, Natural History, and Practice Patterns of Patients with Myelodysplastic Syndromes in 2010. J. Natl. Compr. Cancer Netw. 2011, 9, 57–63.
- Cogle, C.R.; Craig, B.M.; Rollison, D.E.; List, A.F. Incidence of the Myelodysplastic Syndromes Using a Novel Claims-Based Algorithm: High Number of Uncaptured Cases by Cancer Registries. Blood 2011, 117, 7121–7125.
- Germing, U.; Aul, C.; Niemeyer, C.M.; Haas, R.; Bennett, J.M. Epidemiology, Classification and Prognosis of Adults and Children with Myelodysplastic Syndromes. Ann. Hematol. 2008, 87, 691–699.
- Efficace, F.; Gaidano, G.; Breccia, M.; Criscuolo, M.; Cottone, F.; Caocci, G.; Bowen, D.; Lübbert, M.; Angelucci, E.; Stauder, R.; et al. Prevalence, Severity and Correlates of Fatigue in Newly Diagnosed Patients with Myelodysplastic Syndromes. Br. J. Haematol. 2015, 168, 361–370.
- Pomeroy, C.; Oken, M.; Rydell, R.E.; Filice, G.A. Infection in the Myelodysplastic Syndromes. Am. J. Med. 1991, 90, 338–344.
- Anderson, L.A.; Pfeiffer, R.M.; Landgren, O.; Gadalla, S.; Berndt, S.I.; Engels, E.A. Risks of Myeloid Malignancies in Patients with Autoimmune Conditions. Br. J. Cancer 2009, 100, 822–828.
- Steensma, D.P.; Bennett, J.M. The Myelodysplastic Syndromes: Diagnosis and Treatment. Mayo Clin. Proc. 2006, 81, 104–130.
- Swerdlow, S.H.; Campo, E.; Harris, N.L.; Jaffe, E.S.; Pileri, S.A.; Stein, H.; Thiele, J. WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues, 4th ed.; International Agency for Research on Cancer: Lyon, France, 2017; Volume 2, p. 188.
- Papaemmanuil, E.; Gerstung, M.; Bullinger, L.; Gaidzik, V.I.; Paschka, P.; Roberts, N.D.; Potter, N.E.; Heuser, M.; Thol, F.; Bolli, N.; et al. Genomic Classification and Prognosis in Acute Myeloid Leukemia. N. Engl. J. Med. 2016, 374, 2209–2221.
- Lindsley, R.C.; Mar, B.G.; Mazzola, E.; Grauman, P.V.; Shareef, S.; Allen, S.L.; Pigneux, A.; Wetzler, M.; Stuart, R.K.; Erba, H.P.; et al. Acute Myeloid Leukemia Ontogeny Is Defined by Distinct Somatic Mutations. Blood 2015, 125, 1367–1376.
- Lane, S.W.; Scadden, D.T.; Gilliland, D.G. The Leukemic Stem Cell Niche: Current Concepts and Therapeutic Opportunities. Blood 2009, 114, 1150–1157.
- Pronk, E.; Raaijmakers, M.H.G.P. The Mesenchymal Niche in MDS. Blood 2019, 133, 1031–1038.
- Papaemmanuil, E.; Gerstung, M.; Malcovati, L.; Tauro, S.; Gundem, G.; Van Loo, P.; Yoon, C.J.; Ellis, P.; Wedge, D.C.; Pellagatti, A.; et al. Clinical and Biological Implications of Driver Mutations in Myelodysplastic Syndromes. Blood 2013, 122, 3616–3627, quiz 3699.
- Abdel-Wahab, O.; Adli, M.; LaFave, L.M.; Gao, J.; Hricik, T.; Shih, A.H.; Pandey, S.; Patel, J.P.; Chung, Y.R.; Koche, R.; et al. ASXL1 Mutations Promote Myeloid Transformation through Loss of PRC2-Mediated Gene Repression. Cancer Cell 2012, 22, 180–193.
- Schnittger, S.; Eder, C.; Jeromin, S.; Alpermann, T.; Fasan, A.; Grossmann, V.; Kohlmann, A.; Illig, T.; Klopp, N.; Wichmann, H.-E.; et al. ASXL1 Exon 12 Mutations Are Frequent in AML with Intermediate Risk Karyotype and Are Independently Associated with an Adverse Outcome. Leukemia 2013, 27, 82–91.
- Metzeler, K.H.; Becker, H.; Maharry, K.; Radmacher, M.D.; Kohlschmidt, J.; Mrózek, K.; Nicolet, D.; Whitman, S.P.; Wu, Y.-Z.; Schwind, S.; et al. ASXL1 Mutations Identify a High-Risk Subgroup of Older Patients with Primary Cytogenetically Normal AML within the ELN Favorable Genetic Category. Blood 2011, 118, 6920–6929.
- Boultwood, J.; Perry, J.; Pellagatti, A.; Fernandez-Mercado, M.; Fernandez-Santamaria, C.; Calasanz, M.J.; Larrayoz, M.J.; Garcia-Delgado, M.; Giagounidis, A.; Malcovati, L.; et al. Frequent Mutation of the Polycomb-Associated Gene ASXL1 in the Myelodysplastic Syndromes and in Acute Myeloid Leukemia. Leukemia 2010, 24, 1062–1065.
- Sashida, G.; Oshima, M.; Iwama, A. Deregulated Polycomb Functions in Myeloproliferative Neoplasms. Int. J. Hematol. 2019, 110, 170–178.
- Damm, F.; Chesnais, V.; Nagata, Y.; Yoshida, K.; Scourzic, L.; Okuno, Y.; Itzykson, R.; Sanada, M.; Shiraishi, Y.; Gelsi-Boyer, V.; et al. BCOR and BCORL1 Mutations in Myelodysplastic Syndromes and Related Disorders. Blood 2013, 122, 3169–3177.
- Kelly, M.J.; So, J.; Rogers, A.J.; Gregory, G.; Li, J.; Zethoven, M.; Gearhart, M.D.; Bardwell, V.J.; Johnstone, R.W.; Vervoort, S.J.; et al. Bcor Loss Perturbs Myeloid Differentiation and Promotes Leukaemogenesis. Nat. Commun. 2019, 10, 1347.
- Russler-Germain, D.A.; Spencer, D.H.; Young, M.A.; Lamprecht, T.L.; Miller, C.A.; Fulton, R.; Meyer, M.R.; Erdmann-Gilmore, P.; Townsend, R.R.; Wilson, R.K.; et al. The R882H DNMT3A Mutation Associated with AML Dominantly Inhibits Wild-Type DNMT3A by Blocking Its Ability to Form Active Tetramers. Cancer Cell 2014, 25.
- Challen, G.A.; Sun, D.; Jeong, M.; Luo, M.; Jelinek, J.; Berg, J.S.; Bock, C.; Vasanthakumar, A.; Gu, H.; Xi, Y.; et al. Dnmt3a Is Essential for Hematopoietic Stem Cell Differentiation. Nat. Genet. 2012, 44.
- Ettou, S.; Audureau, E.; Humbrecht, C.; Benet, B.; Jammes, H.; Clozel, T.; Bardet, V.; Lacombe, C.; Dreyfus, F.; Mayeux, P.; et al. Fas Expression at Diagnosis as a Biomarker of Azacitidine Activity in High-Risk MDS and Secondary AML. Leukemia 2012, 26, 2297–2299.
- Shlush, L.I.; Zandi, S.; Mitchell, A.; Chen, W.C.; Brandwein, J.M.; Gupta, V.; Kennedy, J.A.; Schimmer, A.D.; Schuh, A.C.; Yee, K.W.; et al. Identification of Pre-Leukaemic Haematopoietic Stem Cells in Acute Leukaemia. Nature 2014, 506, 328–333.
- Welch, J.S.; Ley, T.J.; Link, D.C.; Miller, C.A.; Larson, D.E.; Koboldt, D.C.; Wartman, L.D.; Lamprecht, T.L.; Liu, F.; Xia, J.; et al. The Origin and Evolution of Mutations in Acute Myeloid Leukemia. Cell 2012, 150, 264–278.
- Ding, L.; Ley, T.J.; Larson, D.E.; Miller, C.A.; Koboldt, D.C.; Welch, J.S.; Ritchey, J.K.; Young, M.A.; Lamprecht, T.; Mclellan, M.D.; et al. Clonal Evolution in Relapsed Acute Myeloid Leukaemia Revealed by Whole-Genome Sequencing. Nature 2012.
- Herviou, L.; Cavalli, G.; Cartron, G.; Klein, B.; Moreaux, J. EZH2 in Normal Hematopoiesis and Hematological Malignancies. Oncotarget 2016, 7, 2284–2296.
- Lund, K.; Adams, P.D.; Copland, M. EZH2 in Normal and Malignant Hematopoiesis. Leukemia 2014, 28, 44–49.
- Nikoloski, G.; Langemeijer, S.M.C.; Kuiper, R.P.; Knops, R.; Massop, M.; Tönnissen, E.R.L.T.M.; van der Heijden, A.; Scheele, T.N.; Vandenberghe, P.; de Witte, T.; et al. Somatic Mutations of the Histone Methyltransferase Gene EZH2 in Myelodysplastic Syndromes. Nat. Genet. 2010, 42, 665–667.
- Sashida, G.; Harada, H.; Matsui, H.; Oshima, M.; Yui, M.; Harada, Y.; Tanaka, S.; Mochizuki-Kashio, M.; Wang, C.; Saraya, A.; et al. Ezh2 Loss Promotes Development of Myelodysplastic Syndrome but Attenuates Its Predisposition to Leukaemic Transformation. Nat. Commun. 2014, 5, 4177.
- Neff, T.; Sinha, A.U.; Kluk, M.J.; Zhu, N.; Khattab, M.H.; Stein, L.; Xie, H.; Orkin, S.H.; Armstrong, S.A. Polycomb Repressive Complex 2 Is Required for MLL-AF9 Leukemia. Proc. Natl. Acad. Sci. USA 2012, 109, 5028–5033.
- Medeiros, B.C.; Fathi, A.T.; DiNardo, C.D.; Pollyea, D.A.; Chan, S.M.; Swords, R. Isocitrate Dehydrogenase Mutations in Myeloid Malignancies. Leukemia 2017, 31, 272–281.
- Xu, W.; Yang, H.; Liu, Y.; Yang, Y.; Wang, P.; Kim, S.-H.; Ito, S.; Yang, C.; Wang, P.; Xiao, M.-T.; et al. Oncometabolite 2-Hydroxyglutarate Is a Competitive Inhibitor of α-Ketoglutarate-Dependent Dioxygenases. Cancer Cell 2011, 19, 17–30.
- Figueroa, M.E.; Abdel-Wahab, O.; Lu, C.; Ward, P.S.; Patel, J.; Shih, A.; Li, Y.; Bhagwat, N.; Vasanthakumar, A.; Fernandez, H.F.; et al. Leukemic IDH1 and IDH2 Mutations Result in a Hypermethylation Phenotype, Disrupt TET2 Function, and Impair Hematopoietic Differentiation. Cancer Cell 2010, 18, 553–567.
- Lu, C.; Ward, P.S.; Kapoor, G.S.; Rohle, D.; Turcan, S.; Abdel-Wahab, O.; Edwards, C.R.; Khanin, R.; Figueroa, M.E.; Melnick, A.; et al. IDH Mutation Impairs Histone Demethylation and Results in a Block to Cell Differentiation. Nature 2012, 483, 474–478.
- DiNardo, C.D.; Jabbour, E.; Ravandi, F.; Takahashi, K.; Daver, N.; Routbort, M.; Patel, K.P.; Brandt, M.; Pierce, S.; Kantarjian, H.; et al. IDH1 and IDH2 Mutations in Myelodysplastic Syndromes and Role in Disease Progression. Leukemia 2016, 30, 980–984.
- Calvert, A.E.; Chalastanis, A.; Wu, Y.; Hurley, L.A.; Kouri, F.M.; Bi, Y.; Kachman, M.; May, J.L.; Bartom, E.; Hua, Y.; et al. Cancer-Associated IDH1 Promotes Growth and Resistance to Targeted Therapies in the Absence of Mutation. Cell Rep. 2017, 19, 1858–1873.
- De Braekeleer, E.; Douet-Guilbert, N.; Morel, F.; Le Bris, M.-J.; Férec, C.; De Braekeleer, M. RUNX1 Translocations and Fusion Genes in Malignant Hemopathies. Future Oncol. Lond. Engl. 2011, 7, 77–91.
- Osato, M. Point Mutations in the RUNX1/AML1 Gene: Another Actor in RUNX Leukemia. Oncogene 2004, 23, 4284–4296.
- Harada, H.; Harada, Y.; Niimi, H.; Kyo, T.; Kimura, A.; Inaba, T. High Incidence of Somatic Mutations in the AML1/RUNX1 Gene in Myelodysplastic Syndrome and Low Blast Percentage Myeloid Leukemia with Myelodysplasia. Blood 2004, 103, 2316–2324.
- Jacob, B.; Osato, M.; Yamashita, N.; Wang, C.Q.; Taniuchi, I.; Littman, D.R.; Asou, N.; Ito, Y. Stem Cell Exhaustion Due to Runx1 Deficiency Is Prevented by Evi5 Activation in Leukemogenesis. Blood 2010, 115, 1610–1620.
- Huang, G.; Zhang, P.; Hirai, H.; Elf, S.; Yan, X.; Chen, Z.; Koschmieder, S.; Okuno, Y.; Dayaram, T.; Growney, J.D.; et al. PU.1 Is a Major Downstream Target of AML1 (RUNX1) in Adult Mouse Hematopoiesis. Nat. Genet. 2008, 40, 51–60.
- Yu, M.; Mazor, T.; Huang, H.; Huang, H.-T.; Kathrein, K.L.; Woo, A.J.; Chouinard, C.R.; Labadorf, A.; Akie, T.E.; Moran, T.B.; et al. Direct Recruitment of Polycomb Repressive Complex 1 to Chromatin by Core Binding Transcription Factors. Mol. Cell 2012, 45, 330–343.
- Nasmyth, K.; Haering, C.H. Cohesin: Its Roles and Mechanisms. Annu. Rev. Genet. 2009, 43, 525–558.
- Van Ruiten, M.S.; Rowland, B.D. On the Choreography of Genome Folding: A Grand Pas de Deux of Cohesin and CTCF. Curr. Opin. Cell Biol. 2021, 70, 84–90.
- Thota, S.; Viny, A.D.; Makishima, H.; Spitzer, B.; Radivoyevitch, T.; Przychodzen, B.; Sekeres, M.A.; Levine, R.L.; Maciejewski, J.P. Genetic Alterations of the Cohesin Complex Genes in Myeloid Malignancies. Blood 2014, 124, 1790–1798.
- Smith, J.S.; Lappin, K.M.; Craig, S.G.; Liberante, F.G.; Crean, C.M.; McDade, S.S.; Thompson, A.; Mills, K.I.; Savage, K.I. Chronic Loss of STAG2 Leads to Altered Chromatin Structure Contributing to De-Regulated Transcription in AML. J. Transl. Med. 2020, 18, 339.
- Cuartero, S.; Weiss, F.D.; Dharmalingam, G.; Guo, Y.; Ing-Simmons, E.; Masella, S.; Robles-Rebollo, I.; Xiao, X.; Wang, Y.-F.; Barozzi, I.; et al. Control of Inducible Gene Expression Links Cohesin to Hematopoietic Progenitor Self-Renewal and Differentiation. Nat. Immunol. 2018, 19, 932–941.
- Tothova, Z.; Valton, A.-L.; Gorelov, R.A.; Vallurupalli, M.; Krill-Burger, J.M.; Holmes, A.; Landers, C.C.; Haydu, J.E.; Malolepsza, E.; Hartigan, C.; et al. Cohesin Mutations Alter DNA Damage Repair and Chromatin Structure and Create Therapeutic Vulnerabilities in MDS/AML. JCI Insight 2021, 6.
- López-Moyado, I.F.; Rao, A. DNMT3A and TET2 Mutations Reshape Hematopoiesis in Opposing Ways. Nat. Genet. 2020, 52.
- Nakajima, H.; Kunimoto, H. TET2 as an Epigenetic Master Regulator for Normal and Malignant Hematopoiesis. Cancer Sci. 2014, 105, 1093–1099.
- Jankowska, A.M.; Szpurka, H.; Tiu, R.V.; Makishima, H.; Afable, M.; Huh, J.; O’Keefe, C.L.; Ganetzky, R.; McDevitt, M.A.; Maciejewski, J.P. Loss of Heterozygosity 4q24 and TET2 Mutations Associated with Myelodysplastic/Myeloproliferative Neoplasms. Blood 2009, 113, 6403–6410.
- Rasmussen, K.D.; Jia, G.; Johansen, J.V.; Pedersen, M.T.; Rapin, N.; Bagger, F.O.; Porse, B.T.; Bernard, O.A.; Christensen, J.; Helin, K. Loss of TET2 in Hematopoietic Cells Leads to DNA Hypermethylation of Active Enhancers and Induction of Leukemogenesis. Genes Dev. 2015, 29.
- Itzykson, R.; Kosmider, O.; Renneville, A.; Morabito, M.; Preudhomme, C.; Berthon, C.; Adès, L.; Fenaux, P.; Platzbecker, U.; Gagey, O.; et al. Clonal Architecture of Chronic Myelomonocytic Leukemias. Blood 2013, 121.
- Abdel-Wahab, O.; Mullally, A.; Hedvat, C.; Garcia-Manero, G.; Patel, J.; Wadleigh, M.; Malinge, S.; Yao, J.; Kilpivaara, O.; Bhat, R.; et al. Genetic Characterization of TET1, TET2, and TET3 Alterations in Myeloid Malignancies. Blood 2009, 114, 144–147.
- Kosmider, O.; Gelsi-Boyer, V.; Cheok, M.; Grabar, S.; Della-Valle, V.; Picard, F.; Viguié, F.; Quesnel, B.; Beyne-Rauzy, O.; Solary, E.; et al. TET2 Mutation Is an Independent Favorable Prognostic Factor in Myelodysplastic Syndromes (MDSs). Blood 2009, 114, 3285–3291.
- Tefferi, A.; Levine, R.L.; Lim, K.-H.; Abdel-Wahab, O.; Lasho, T.L.; Patel, J.; Finke, C.M.; Mullally, A.; Li, C.-Y.; Pardanani, A.; et al. Frequent TET2 Mutations in Systemic Mastocytosis: Clinical, KITD816V and FIP1L1-PDGFRA Correlates. Leukemia 2009, 23, 900–904.
- Ji, H.; Ehrlich, L.I.R.; Seita, J.; Murakami, P.; Doi, A.; Lindau, P.; Lee, H.; Aryee, M.J.; Irizarry, R.A.; Kim, K.; et al. Comprehensive Methylome Map of Lineage Commitment from Haematopoietic Progenitors. Nature 2010, 467, 338–342.
- Trowbridge, J.J.; Snow, J.W.; Kim, J.; Orkin, S.H. DNA Methyltransferase 1 Is Essential for and Uniquely Regulates Hematopoietic Stem and Progenitor Cells. Cell Stem Cell 2009, 5, 442–449.
- Bröske, A.-M.; Vockentanz, L.; Kharazi, S.; Huska, M.R.; Mancini, E.; Scheller, M.; Kuhl, C.; Enns, A.; Prinz, M.; Jaenisch, R.; et al. DNA Methylation Protects Hematopoietic Stem Cell Multipotency from Myeloerythroid Restriction. Nat. Genet. 2009, 41, 1207–1215.
- Jiang, Y.; Dunbar, A.; Gondek, L.P.; Mohan, S.; Rataul, M.; O’Keefe, C.; Sekeres, M.; Saunthararajah, Y.; Maciejewski, J.P. Aberrant DNA Methylation Is a Dominant Mechanism in MDS Progression to AML. Blood 2009, 113.
- Yang, X.; Wong, M.P.M.; Ng, R.K. Aberrant DNA Methylation in Acute Myeloid Leukemia and Its Clinical Implications. Int. J. Mol. Sci. 2019, 20, 4576.
- Valk-Lingbeek, M.E.; Bruggeman, S.W.M.; van Lohuizen, M. Stem Cells and Cancer. Cell 2004, 118, 409–418.
- Trojer, P. Chapter 37: Histone Methylation Modifiers in Medical Therapeutics. In Medical Epigenetics; Academic Press: Cambridge, MA, USA, 2016; pp. 705–729.
- Sashida, G.; Iwama, A. Epigenetic Regulation of Hematopoiesis. Int. J. Hematol. 2012, 96, 405–412.
- Bernstein, B.E.; Mikkelsen, T.S.; Xie, X.; Kamal, M.; Huebert, D.J.; Cuff, J.; Fry, B.; Meissner, A.; Wernig, M.; Plath, K.; et al. A Bivalent Chromatin Structure Marks Key Developmental Genes in Embryonic Stem Cells. Cell 2006, 125, 315–326.
- Cedar, H.; Bergman, Y. Epigenetics of Haematopoietic Cell Development. Nat. Rev. Immunol. 2011, 11, 478–488.
- Weishaupt, H.; Sigvardsson, M.; Attema, J.L. Epigenetic Chromatin States Uniquely Define the Developmental Plasticity of Murine Hematopoietic Stem Cells. Blood 2010, 115, 247–256.
- Bejar, R.; Stevenson, K.; Abdel-Wahab, O.; Galili, N.; Nilsson, B.; Garcia-Manero, G.; Kantarjian, H.; Raza, A.; Levine, R.L.; Neuberg, D.; et al. Clinical Effect of Point Mutations in Myelodysplastic Syndromes. N. Engl. J. Med. 2011, 364, 2496–2506.
- Wang, Z.; Gearhart, M.D.; Lee, Y.-W.; Kumar, I.; Ramazanov, B.; Zhang, Y.; Hernandez, C.; Lu, A.Y.; Neuenkirchen, N.; Deng, J.; et al. A Non-Canonical BCOR-PRC1.1 Complex Represses Differentiation Programs in Human ESCs. Cell Stem Cell 2018, 22.
- Kandoth, C.; McLellan, M.D.; Vandin, F.; Ye, K.; Niu, B.; Lu, C.; Xie, M.; Zhang, Q.; McMichael, J.F.; Wyczalkowski, M.A.; et al. Mutational Landscape and Significance across 12 Major Cancer Types. Nature 2013, 502, 333–339.

