Mutations in genes encoding chromatin regulators are early events contributing to developing asymptomatic clonal hematopoiesis of indeterminate potential and its frequent progression to myeloid diseases with increasing severity. We focus on the subset of myeloid diseases encompassing myelodysplastic syndromes and their transformation to secondary acute myeloid leukemia. We introduce the major concepts of chromatin regulation that provide the basis of epigenetic regulation. In greater detail, we discuss those chromatin regulators that are frequently mutated in myelodysplastic syndromes. We discuss their role in the epigenetic regulation of normal hematopoiesis and the consequence of their mutation. Finally, we provide an update on the drugs interfering with chromatin regulation approved or in development for myelodysplastic syndromes and acute myeloid leukemia.
- epigenetics
- chromatin
- epigenetic regulators
- clonal hematopoiesis of indeterminate potential (CHIP)
- myelodysplastic syndromes (MDS)
- acute myeloid leukemia (AML)
- secondary acute myeloid leukemia (sAML)
1. Introduction
Myelodysplastic syndromes (MDS) are part of a spectrum of clonal myeloid diseases starting with the asymptomatic expansion of mutated hematopoietic stem cell (HSC) clones and frequently ending with transformation to full-blown secondary acute myeloid leukemia (sAML) [1]. The evolution and progression of MDS and sAML is intimately linked to changes in the regulation of chromatin function and epigenetics. First, effector enzymes with epigenetic regulatory functions are among the most commonly mutated genes in MDS and AML [2,3]. Second, epigenetic abnormalities co-occur with genetic and cytogenetic changes in MDS and sAML, and together, contribute to the full manifestation of the disease [4]. Indeed, the accumulation of epigenetic changes has been suggested to represent a tipping point to transformation to sAML [1]. The fact that epigenetic changes are reversible has provided the rationale for developing therapies that target epigenetic regulators.
2. CHIP-MDS-sAML—A Spectrum Myeloid Diseases
The expansion of clonal populations of blood cells from a single hematopoietic stem cell (HSC) with one or more somatic mutations is divided into two categories age-related clonal hematopoiesis (ARCH) and clonal hematopoiesis of indeterminate potential (CHIP). ARCH describes broad recurrently occurring mutational events that can cause clonal hematopoiesis and lead to age-related pathologies, including inflammation, cancer mortality, as well as hematological malignancies [6]. On the other hand, CHIP is associated with detectable somatic clonal mutations in leukemia-driver genes with a variant allele frequency (VAF) of 2% or greater [8] (Figure 1). Individuals with CHIP show normal peripheral blood counts and no evidence of WHO-defined criteria for a hematological malignancy or other clonal disorders [9]. Mutations that also occur in MDS and sAML have been observed in healthy, mainly elderly populations as part of population-based studies [10,11]. CHIP-related mutational burden appears to increase with age, as CHIP is present in 10–15% of individuals aged over 70 years [1]. Interestingly, the most frequent mutations in CHIP affect the epigenetic regulators TET2, DNMT3A and ASXL1 and the splicing factor SF3B1. Individuals with CHIP have an increased risk of developing diseases of the lymphoid and myeloid lineage, including MDS. This happens when mutations increase the fitness of HSC clones allowing them to expand among the bulk HSC population, eventually resulting in clonal dominance. If mutations are coupled with reduced differentiation capacity, the expansion of mutated HSCs can lead to reduced generation of mature blood cells in one or several lineages (Figure 1). The current challenge lies in understanding how CHIP predisposes to developing disorders. For a more thorough discussion of CHIP and its consequences, please see recent reviews [6,8].
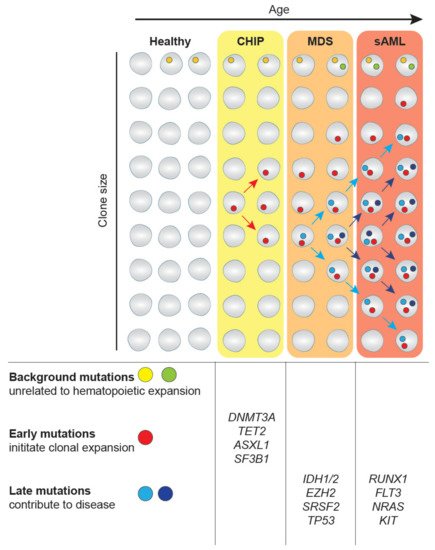
MDS is the most frequent hematopoietic disorder in the elderly [12,13]. Advanced age is the main contributing risk factor of acute myeloid malignancies, with the median age of diagnosis at around 70 years and 92% of MDS patients aged over 50 years [14,15]. MDS is characterized by the expansion of mutant HSC clones at the expense of normal hematopoiesis leading to low blast cell counts, but a substantial reduction of numbers of mature blood cell types referred to as cytopenias. Consequential symptoms are fatigue due to anemia [16], recurring infections related to neutrophil dysfunction [17] and autoimmune abnormalities, such as rheumatic heart disease [18].
Around 30% of MDS patients transform to sAML [19], which is characterized by further increases in blast cell counts above 20% in the bone marrow [20]. On the genetic and molecular level, sAML mutant HSC clones have acquired additional driver mutations that convert them into full leukemia stem cells (LSCs). These genetic alterations differ to some extent from other AML subtypes [21]. De novo AML occurs without any previous neoplasm, is more common in younger patients and is associated with better overall survival [22]. Compared to CHIP and early-stage MDS, LSCs in sAML and late-stage MDS have acquired mutations that confer uncontrolled growth, such as NRAS, and inhibition of apoptosis, such as TP53. Together with epigenetic abnormalities, these oncogenic mutations cause blast cell numbers to increase and inhibit differentiation, which is characteristic of the MDS-to-sAML transformation [1]. Furthermore, an abnormal stem cell niche in the bone marrow may favor the outgrowth of mutant clones and thus contribute to the disease [23,24].
In summary, MDS and sAML are part of a spectrum of clonal diseases affecting the myeloid lineage that can arise from CHIP. Mutations in epigenetic regulators are early events and provide a yet not fully understood function in disease etiology.
3. Epigenetic Regulators Frequently Mutated in Myeloid Diseases and Their Function
Recurrent mutations in CHIP, MDS and sAML affecting genes involved in epigenetic regulation include regulators of DNA methylation, histone modifiers and elements regulating higher-order chromatin architecture [2,41]. For these groups of genes, we discuss their normal role in hematopoiesis and the consequences of their mutations in the disease (summarized in Table 1). Again, we focus on MDS and sAML but also discuss selected insights from other types of AML.
| Gene | Mutation Effect on Gene | Mutational Frequency | Characteristics |
|---|---|---|---|
| ASXL1 [42,43,44,45] | Loss-of-function mutation | 20% in MDS | Mutations enriched in elderly AML and sAML patients |
| 6–30% in AML | |||
| BCOR [46,47,48] | Loss-of-function mutation | 5% in MDS | Associated with poor prognosis |
| 9% in AML | |||
| DNMT3A [49,50,51,52,53,54] | Loss-of-function mutation | 13% in MDS | Thought to be initiating mutation during the pre-leukemic state |
| 20% in AML | Important for the balance of differentiation and self-renewal | ||
| EZH2 [55,56,57,58,59] | Loss-of-function mutation as well as gain of function mutations | 5% in MDS | Thought to regulate the balance between self-renewal and differentiation |
| 1–2% de novo AML | In MDS associated with poor prognosis | ||
| IDH1/2 [60,61,62,63,64,65] | Gain of function | 5% in MDS | Leads to the production of oncometabolite, which interferes with TET2 activity and histone demethylases |
| 20% in AML | IDH2 mutations are more common | ||
| RUNX1 [66,67,68,69,70,71] | Translocations | 10–20% in MDS | Significantly associated with EZH2 mutations |
| Loss-of-function mutation | 2–20% in AML | ||
| Cohesin [72,73,74,75,76,77] | Loss-of-function mutation | 10–15% in MDS, | Mutually exclusive |
| 10% in AML | often associated with mutations in NPM1, TET2, ASXL1 and EZH2 | ||
| TET2 [78,79,80,81,82,83,84,85] | Loss-of-function mutation | 30–50% in MDS | Important for myeloid differentiation and lineage commitment |
| 30% in sAML | Associated with poor prognosis in some studies |
3.1. Mutations Causing Aberrant DNA Methylation—TET2, DNMT3A, IDH
3.2. Dysregulation of Histone Modifications—EZH2, RUNX1, BCOR, ASXL1
3.3. Altering Chromatin Structure—The Cohesin Complex
Somatic mutations affecting the cohesin complex have been identified in several diseases, including MDS and AML [98]. The cohesin complex consists of the core subunits SMC1, SMC3 and RAD21, which associate with either STAG1 or STAG2. One of its important functions is to align and stabilize sister chromatids during metaphase crucial for DNA replication, DNA repair and mitosis [72]. In addition, cohesin has an important role in the regulation of genome folding in interphase cells [73]. Loss-of-function cohesin mutations, mainly in the STAG2 gene, were detected in 10–15% of MDS and 20% of sAML patients and are associated with poor survival [74]. Interestingly, in several human leukemic cell lines, low expression of cohesin was observed, although no mutation could be identified [74]. On the mechanistic level, reduced cohesin function leads to changes in gene expression, possibly as a direct consequence of changes in chromatin architecture [75]. In particular, reduced sensitivity to inflammatory signals may affect the function of HSCs [76].
In conclusion, with mutations affecting cohesin, histone-modifying PRCs and the DNA methylation machinery, several central epigenetic mechanisms are perturbed in MDS and sAML. The common denominator of these mutations in disease is that they disrupt normal hematopoietic differentiation and promote the expansion of altered HSCs [74], thereby contributing to disease progression. The challenge for the field now is to identify specific vulnerabilities of mutant cells that can be exploited for therapeutic strategies aiming at synthetic lethality. An exciting example is a recent demonstration that cohesin mutant cells are hypersensitive to inhibitors of the DNA repair pathway [77].
This entry is adapted from the peer-reviewed paper 10.3390/cancers13071746