 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Hongmei Du | + 1091 word(s) | 1091 | 2021-04-06 06:09:54 | | | |
| 2 | Vivi Li | + 11 word(s) | 1102 | 2021-04-13 10:20:35 | | | | |
| 3 | Vivi Li | + 11 word(s) | 1102 | 2021-04-13 10:21:29 | | | | |
| 4 | Vivi Li | + 11 word(s) | 1102 | 2021-04-13 10:22:44 | | |
Video Upload Options
Purslane (Portulaca oleracea) first came from India and Iran and has spread around the world. It is a warm-climate, juicy annual plant spread around the world, belonging to the Portulacaceae family.
1. Introduction
Purslane (Portulaca oleracea) first came from India and Iran and has spread around the world. It is a warm-climate, juicy annual plant spread around the world, belonging to the Portulacaceae family. Purslane is one of the most abundant terrestrial vegetables in spite of its genetic assortment [1][2][3] and generally recognized as purslane (the USA and Australia), Ma-Chi-Xian (China) [4] and kurfa in (Pakistan) [5][6]. It has been used as an important traditional medicinal plant and itemized by the World Health Organization as “Global Panacea” [7].
Apart from medicinal and nutritive value, purslane is a high-quality halophyte [8]. Some results have been reported about its salt tolerance mechanism [9][10][11][12]. Since, the development of high-throughput sequencing technology, transcripts sequencing has become an important means to study gene expression regulation, after the whole-genome sequencing of human were completed in 2004 [13]. However, due to the read length limitation of the second-generation sequencing in different organisms [14][15][16][17][18], the full-length transcript obtained by splicing is not complete. The third-generation sequencing technology signified by Pacific Biosciences (PacBio, Menlo Park, CA, USA) effectively overcome this problem [19][20]. Single-molecule real-time (SMRT) sequencing can directly obtain full-length splice isoforms without the need for assembly [21][22], improve the draft genome annotation in species with reference genome and facilitate comparative transcriptome studies and gene functional annotation [22]. Up to date, this technology has been successfully utilized in some species, such as perennial ryegrass (Lolium perenne) [22], Rhododendron lapponicum [23], strawberry (Fragaria × ananassa) [24], Gnetum luofuense [25], and maize (Zea mays) [21]. However, up to date, no researcher studied the full-length transcript of purslane using single-molecule long-read sequencing. In this study, SMRT sequencing was achieved in purslane. After detecting transcripts, we completed functional annotations of transcripts, transcript factors (TFs) and simple sequence repeat (SSR) analysis, long non-coding RNAs (LncRNA) prediction. This study established a high-quality reference transcriptome for purslane, which provides valuable resources for further investigation of related molecular mechanisms, especially biosynthesis of unsaturated fatty acids pathway in purslane.
2. Single-Molecule Real-Time Sequencing of Purslane
Leaves and roots from “Pakistan local” (“PL”-North American origin) and a wild variety “Liaoning, China local” (“LCL”) were used for RNA extraction and cDNA library construction. After removing adaptor sequences, low-quality sequences, and short sequences less than 50 bp, a total of 9,350,222 subread (15.33 Gb) were obtained, through normal subread length of 1640 bp, and N50, of 3093 bp (Figure 1A). After self-correction of subread sequences (with min passes = 2, min predicted accuracy = 0.8), a total of 375,102 circular consensus sequence (CCS) were obtained. After sequencing, 259,265 full-length and 254,692 full-length non-chimeric (FLNC) picks were identified. The average FLNC read length was 2808 bp (Figure 1B). The FLNC sequences of the same transcript were clustered using the iterative isoform-clustering (ICE) algorithm, and 132,536 consensus reads were obtained after clustering (Figure 1C).
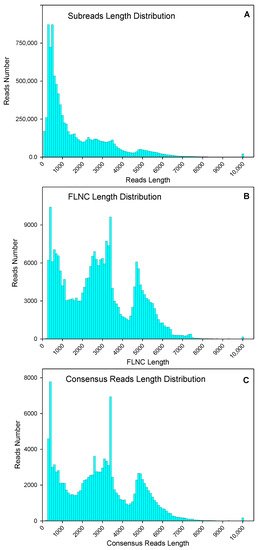
Figure 1. Numbers and length distributions of 9,350,222 subreads (A), 254,692 full-length non-chimeric (FLNC) sequences (B), and 132,536 consensus reads (C) with PacBio single-molecule real-time sequencing method of purslane (Portulaca oleracea).
The number of genes with no isoform remained 53,977, and 78,559 transcriptions were found between 1 and 10 isoforms (Figure 2). The transcript length extended from 170 bp to 14,287 bp, with an average length of 3061 bp.
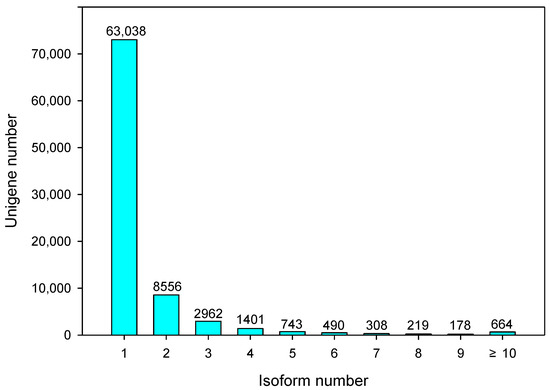
Figure 2. Number of isoforms in purslane (Portulaca oleracea).
3. Functional Annotation of Full-Length Transcripts
All 78,559 unique SMRT transcripts were functionally annotated by searching seven data storage, including Gene Ontology (GO), eukaryotic ortholog groups (KOG), protein family (Pfam), NCBI nucleotide sequences (NT), NCBI nonredundant protein sequences (NR), Swiss-Prot, Kyoto Encyclopedia of Genes and Genomes (KEGG). 90.39% transcript, a total of 71,008, was annotated at one database at least, and 23,013 transcripts were annotated at all seven databases (Figure 3A). We also identified matches to our unique transcripts in clusters of orthologous groups of proteins (COG) (44,376, 56.49%), Pfam database (41,535, 52.87%) and Swiss-Port (58,535, 74.51%). The functional annotations of all 78,559 unique transcripts were listed in Supplementary Table S1. (supplementary could be found in https://www.mdpi.com/2223-7747/10/4/655/htm)
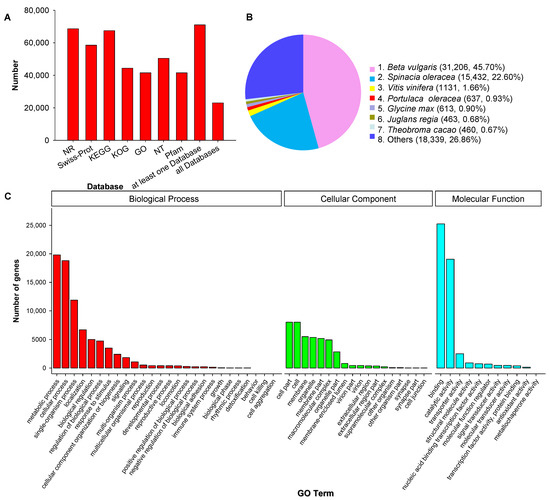
Figure 3. Function annotation of corrected isoforms in seven databases (A) (NR, NCBI Nonredundant Protein Database; KEGG, Kyoto Encyclopedia of Genes and Genomes; KOG, cluster of eukaryotic ortholog groups of proteins; GO, Gene Ontology; NT, NCBI nucleotide sequences; Pfam, Protein family), homologous species distribution diagram of transcripts in NCBI nonredundant protein sequences (NR) (B), Gene Ontology (GO) classification of unique transcripts (C) of purslane (Portulaca oleracea).
We compared the transcript sequences to NR by homologous species analysis. 68,630 genes were annotated, among them, Beta valgaris (31,206; 45.70%), Spinacia oleracea (15,432; 22.60%), Vitis vinifera (1131; 1.66%), and P. oleracea (637; 0.93%) were the top four species of transcripts distributed (Figure 3B). GO analysis demonstrated that 41,535 unique genes were enriched significantly in three major categories: molecular function (MF), cellular component (CC), biological process (BP). In these three categories, the most abundant GO terms were cellular process and metabolic process in BP, catalytic activity and binding in MF, cell and cell part in CC (Figure 3C).
A total of 67,426 sequences were interpreted by the KEGG data storage and plotted to 366 operative catalogs in purslane. “Metabolism” was the largest transcript category. The first three transcripts-related pathways were carbon metabolism (3007, 4.46%), carbon fixation in photosynthetic organisms (2304, 3.42%) and pyruvate metabolism (2153, 3.19%) (Figure 4 and Supplementary Table S1). A large number of genes, especially interrelated in salt-tolerance and the fatty acid component of purslane, were annotated, such as oxidative phosphorylation (l073), plant hormone signal transduction (506), fatty acid biosynthesis (246), biosynthesis of unsaturated fatty acids (94) and α-linolenic acid metabolism (199) (Supplementary Table S1).
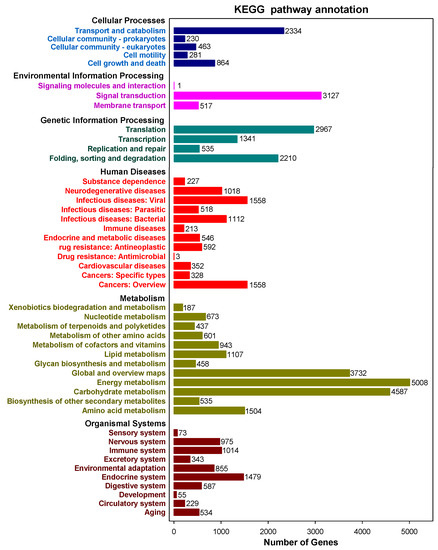
Figure 4. Kyoto Encyclopedia of Genes and Genomes (KEGG) annotated pathways and numbers of the gene in purslane (Portulaca oleracea).
4. Results of Transcript Factors, Long Non-Coding RNAs and Simple Sequence Repeat
During plant growth and development, TFs and transcription regulation (TRs) acting as dominant characters. Four thousand one hundred eighty transcripts, including 2211 putative TF and 1969 TR from 86 families, were predicted with iTAK software [26] (Supplementary Table S2). The top 30 families annotated are shown in Figure 5.
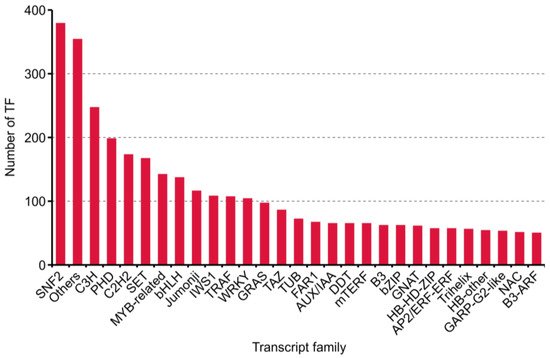
Figure 5. Numbers and families of the top 30 transcript factors (TFs) predicted by iTAK software in purslane (Portulaca oleracea).
LncRNA were predicted by PLEK, Pfam, coding potential calculator (CPC) and coding-non-coding index (CNCI) (Figure 6A). Overall, 7289 LncRNA were predicted with a mean length of 848.84 bp with most LncRNAs length ranging from 300 bp to 1000 bp (Figure 6B and Supplementary Table S3).
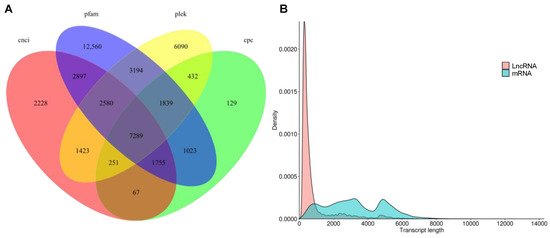
Figure 6. Identification of long non-coding RNAs (LncRNAs). (A). Venn diagram of LncRNAs predicted by PLEK, coding-non-coding index (CNCI), coding potential calculator (CPC) and protein family (Pfam) methods. (B). Density and length distributions of LncRNAs and mRNAs in purslane (Portulaca oleracea).
SSR is also known as short tandem repeats or microsatellite markers. A total of 58,622 sequences were subjected to SSR analysis. Most of the SSRs identified were one-nucleotide repeats (50.81%), followed by compound nucleotide repeats (17.11%), two-nucleotide repeats (15.20%), three-nucleotide repeats (14.76%), four-nucleotide repeats (1.16%), six-nucleotide repeats (0.73%), and five-nucleotide repeats (0.40%) (Table S4).
References
- Ezekwe, M.O.; Omara-Alwala, T.R.; Membrahtu, T. Nutritive characterization of purslane accessions as influenced by planting date. Plant Foods Hum. Nutr. 1999, 54, 183–191.
- Simopoulos, A.P.; Norman, H.A.; Gillaspy, J.E. Purslane in human nutrition and its potential for world agriculture. Plant Foods Hum. Nutr. 1995, 77, 47–74.
- Uddin, M.; Juraimi, A.S.; Hossain, M.S.; Nahar, M.; Ali, M.; Rahman, M. Purslane weed (Portulaca oleracea): A prospective plant source of nutrition, omega-3 fatty acid, and antioxidant attributes. Sci. World J. 2014, 2014, 951019.
- Lee, A.S.; Kim, J.S.; Lee, Y.J.; Kang, D.G.; Lee, H.S. Anti-TNF-α activity of Portulaca oleracea in vascular endothelial cells. Int. J. Mol. Sci. 2012, 13, 5628–5644.
- Hayat, M.Q.; Khan, M.A.; Ahmad, M.; Shaheen, N.; Yasmin, G.; Akhter, S. Ethnotaxonomical approach in the identification of useful medicinal flora of tehsil Pindigheb (District Attock) Pakistan. Ethnobot. Res. Appl. 2008, 6, 35–62.
- Hussain, K.; Nisar, M.F.; Majeed, A.; Nawaz, K.; Bhatti, K.H. Ethnomedicinal survey for important plants of Jalalpur Jattan, district Gujrat, Punjab, Pakistan. Ethnobot. Leafl. 2010, 14, 807–825.
- Ercisli, S.; Coruh, I.; Gormez, A.; Sengul, M. Antioxidant and antibacterial activities of Portulaca oleracea L. Grown wild in Turkey. Ital. J. Food Sci. 2008, 20, 533–542.
- Grieve, C.M.; Suarez, D.L. Purslane (Portulaca oleracea L.): A halophytic crop for drainage water reuse systems. Plant Soil 1997, 192, 277–283.
- Alam, M.A.; Juraimi, A.S.; Rafii, M.Y.; Hamid, A.A. Effect of salinity on biomass yield and physiological and stem-root anatomical characteristics of purslane (Portulaca oleracea L.) accessions. Biomed. Res. Int. 2015, 2015, 105695.
- Kafi, M.; Rahimi, Z. Effect of salinity and silicon on root characteristics, growth, water status, proline content and ion accumulation of purslane (Portulaca oleracea L.). Soil Sci. Plant Nutr. 2011, 57, 341–347.
- Mulry, K.R.; Hanson, B.A.; Dudle, D.A. Alternative strategies in response to saline stress in two varieties of Portulaca oleracea (Purslane). PLoS ONE 2015, 10, e0138723.
- Yazici, I.; Türkan, I.; Sekmen, A.H.; Demiral, T. Salinity tolerance of purslane (Portulaca oleracea L.) is achieved by enhanced antioxidative system, lower level of lipid peroxidation and proline accumulation. Environ. Exp. Bot. 2007, 61, 49–57.
- International Human Genome Sequencing Consortium. Finishing the euchromatic sequence of the human genome. Nature 2004, 431, 931–945.
- Dharshini, S.; Chakravarthi, M.; Manoj, V.; Naveenarani, M.; Kumar, R.; Meena, M.; Ram, B.; Appunu, C. De novo sequencing and transcriptome analysis of a low temperature tolerant Saccharum spontaneum clone IND 00–1037. J. Biotechnol. 2016, 231, 280–294.
- Long, W.; Zou, X.; Zhang, X. Transcriptome analysis of canola (Brassica napus) under salt stress at the germination stage. PLoS ONE 2015, 10, e0116217.
- Li, M.; Liang, Z.; Zeng, Y.; Jing, Y.; Wu, K.; Liang, J.; He, S.; Wang, G.; Mo, Z.; Tan, F. De novo analysis of transcriptome reveals genes associated with leaf abscission in sugarcane (Saccharum officinarum L.). BMC Genom. 2016, 17, 195.
- Wang, J.; Li, B.; Meng, Y.; Ma, X.; Lai, Y.; Si, E.; Yang, K.; Ren, P.; Shang, X.; Wang, H. Transcriptomic profiling of the salt-stress response in the halophyte Halogeton glomeratus. BMC Genom. 2015, 16, 169.
- Xing, J.C.; Zhao, B.Q.; Dong, J.; Liu, C.; Wen, Z.G.; Zhu, X.M.; Ding, H.R.; He, T.T.; Yang, H.; Wang, M.W.; et al. Transcriptome and metabolome profiles revealed molecular mechanisms underlying tolerance of Portulaca oleracea to saline stress. Russ. J. Plant Physiol. 2020, 67, 146–152.
- Jiao, W.B.; Schneeberger, K. The impact of third generation genomic technologies on plant genome assembly. Curr. Opin. Plant Biol. 2017, 36, 64–70.
- Slatko, B.E.; Gardner, A.F.; Ausubel, F.M. Overview of next-generation sequencing technologies. Curr. Protoc. Mol. Biol. 2018, 122, e59.
- Wang, B.; Tseng, E.; Regulski, M.; Clark, T.A.; Hon, T.; Jiao, Y.; Lu, Z.; Olson, A.; Stein, J.C.; Ware, D. Unveiling the complexity of the maize transcriptome by single-molecule long-read sequencing. Nat. Commun. 2016, 7, 11708.
- Xie, L.; Teng, K.; Tan, P.; Chao, Y.; Li, Y.; Guo, W.; Han, L. PacBio single-molecule long-read sequencing shed new light on the transcripts and splice isoforms of the perennial ryegrass. Mol. Genet. Genom. 2020, 295, 475–489.
- Jia, X.; Tang, L.; Mei, X.; Liu, H.; Luo, H.; Deng, Y.; Su, J. Single-molecule long-read sequencing of the full-length transcriptome of Rhododendron lapponicum L. Sci. Rep. 2020, 10, 6755.
- Yuan, H.; Yu, H.; Huang, T.; Shen, X.; Xia, J.; Pang, F.; Wang, J.; Zhao, M. The complexity of the Fragaria × ananassa (octoploid) transcriptome by single-molecule long-read sequencing. Hortic. Res. 2019, 6, 46.
- Deng, N.; Hou, C.; Ma, F.F.; Liu, C.X.; Tian, Y.X. Molecule long-read sequencing reveals the diversity of full-length transcripts in leaves of Gnetum (Gnetales). Int. J. Mol. Sci. 2019, 20, 6350.
- Zheng, Y.; Jiao, C.; Sun, H.; Rosli, H.G.; Pombo, M.A.; Zhang, P.; Banf, M.; Dai, X.; Martin, G.B.; Giovannoni, J.J.; et al. iTAK: A program for genome-wide prediction and classification of plant transcription factors, transcriptional regulators, and protein kinases. Mol. Plant 2016, 9, 1667–1670.

