Purslane (Portulaca oleracea) first came from India and Iran and has spread around the world. It is a warm-climate, juicy annual plant spread around the world, belonging to the Portulacaceae family.
- purslane
- RNA-seq
- gene
- unsaturated fatty acid
1. Introduction
Purslane (Portulaca oleracea) first came from India and Iran and has spread around the world. It is a warm-climate, juicy annual plant spread around the world, belonging to the Portulacaceae family. Purslane is one of the most abundant terrestrial vegetables in spite of its genetic assortment [1,2,3] and generally recognized as purslane (the USA and Australia), Ma-Chi-Xian (China) [4] and kurfa in (Pakistan) [5,6]. It has been used as an important traditional medicinal plant and itemized by the World Health Organization as “Global Panacea” [7].
Apart from medicinal and nutritive value, purslane is a high-quality halophyte [8]. Some results have been reported about its salt tolerance mechanism [9,10,11,12]. Since, the development of high-throughput sequencing technology, transcripts sequencing has become an important means to study gene expression regulation, after the whole-genome sequencing of human were completed in 2004 [13]. However, due to the read length limitation of the second-generation sequencing in different organisms [14,15,16,17,18], the full-length transcript obtained by splicing is not complete. The third-generation sequencing technology signified by Pacific Biosciences (PacBio, Menlo Park, CA, USA) effectively overcome this problem [19,20]. Single-molecule real-time (SMRT) sequencing can directly obtain full-length splice isoforms without the need for assembly [21,22], improve the draft genome annotation in species with reference genome and facilitate comparative transcriptome studies and gene functional annotation [22]. Up to date, this technology has been successfully utilized in some species, such as perennial ryegrass (Lolium perenne) [22], Rhododendron lapponicum [23], strawberry (Fragaria × ananassa) [24], Gnetum luofuense [25], and maize (Zea mays) [21]. However, up to date, no researcher studied the full-length transcript of purslane using single-molecule long-read sequencing. In this study, SMRT sequencing was achieved in purslane. After detecting transcripts, we completed functional annotations of transcripts, transcript factors (TFs) and simple sequence repeat (SSR) analysis, long non-coding RNAs (LncRNA) prediction. This study established a high-quality reference transcriptome for purslane, which provides valuable resources for further investigation of related molecular mechanisms, especially biosynthesis of unsaturated fatty acids pathway in purslane.
2. Single-Molecule Real-Time Sequencing of Purslane
Leaves and roots from “Pakistan local” (“PL”-North American origin) and a wild variety “Liaoning, China local” (“LCL”) were used for RNA extraction and cDNA library construction. After removing adaptor sequences, low-quality sequences, and short sequences less than 50 bp, a total of 9,350,222 subread (15.33 Gb) were obtained, through normal subread length of 1640 bp, and N50, of 3093 bp (Figure 1A). After self-correction of subread sequences (with min passes = 2, min predicted accuracy = 0.8), a total of 375,102 circular consensus sequence (CCS) were obtained. After sequencing, 259,265 full-length and 254,692 full-length non-chimeric (FLNC) picks were identified. The average FLNC read length was 2808 bp (Figure 1B). The FLNC sequences of the same transcript were clustered using the iterative isoform-clustering (ICE) algorithm, and 132,536 consensus reads were obtained after clustering (Figure 1C).
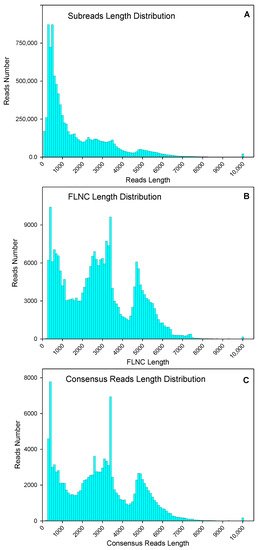
Figure 1. Numbers and length distributions of 9,350,222 subreads (A), 254,692 full-length non-chimeric (FLNC) sequences (B), and 132,536 consensus reads (C) with PacBio single-molecule real-time sequencing method of purslane (Portulaca oleracea).
The number of genes with no isoform remained 53,977, and 78,559 transcriptions were found between 1 and 10 isoforms (Figure 2). The transcript length extended from 170 bp to 14,287 bp, with an average length of 3061 bp.
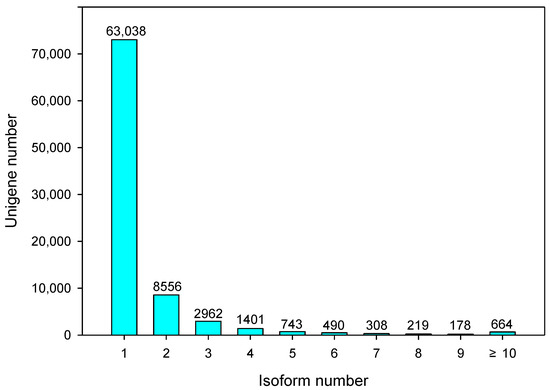
Figure 2. Number of isoforms in purslane (Portulaca oleracea).
3. Functional Annotation of Full-Length Transcripts
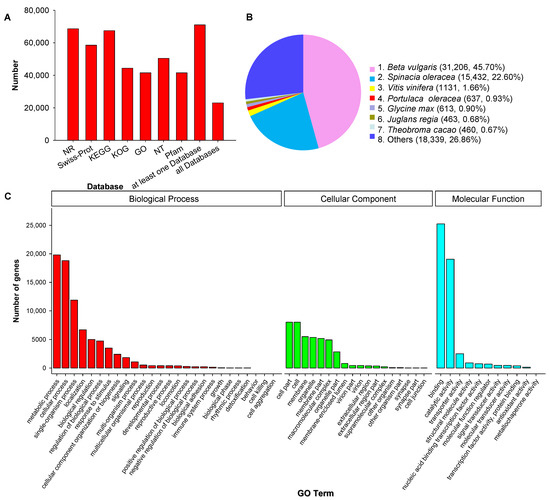
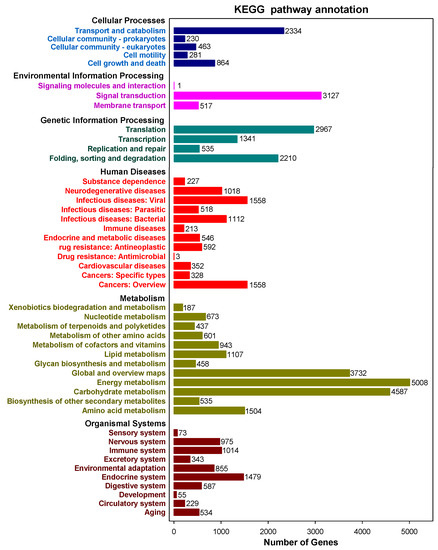
4. Results of Transcript Factors, Long Non-Coding RNAs and Simple Sequence Repeat
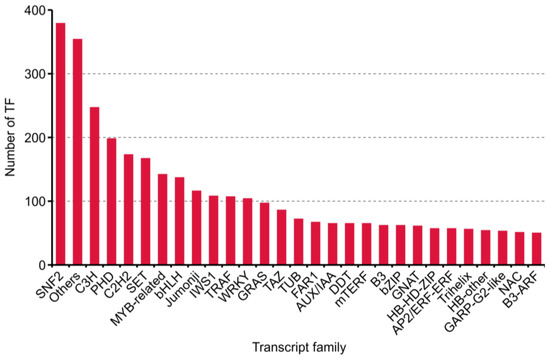
During plant growth and development, TFs and transcription regulation (TRs) acting as dominant characters. Four thousand one hundred eighty transcripts, including 2211 putative TF and 1969 TR from 86 families, were predicted with iTAK software [26] (Supplementary Table S2). The top 30 families annotated are shown in Figure 5.
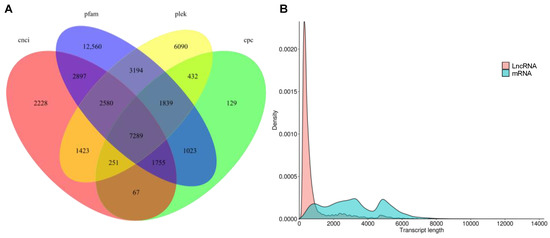
LncRNA were predicted by PLEK, Pfam, coding potential calculator (CPC) and coding-non-coding index (CNCI) (Figure 6A). Overall, 7289 LncRNA were predicted with a mean length of 848.84 bp with most LncRNAs length ranging from 300 bp to 1000 bp (Figure 6B and Supplementary Table S3).
SSR is also known as short tandem repeats or microsatellite markers. A total of 58,622 sequences were subjected to SSR analysis. Most of the SSRs identified were one-nucleotide repeats (50.81%), followed by compound nucleotide repeats (17.11%), two-nucleotide repeats (15.20%), three-nucleotide repeats (14.76%), four-nucleotide repeats (1.16%), six-nucleotide repeats (0.73%), and five-nucleotide repeats (0.40%) (Table S4).
This entry is adapted from the peer-reviewed paper 10.3390/plants10040655