 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Omar Hahad | + 3430 word(s) | 3430 | 2021-04-09 05:23:11 | | | |
| 2 | Camila Xu | Meta information modification | 3430 | 2021-04-15 06:04:42 | | |
Video Upload Options
Aging is a multifactorial dynamic process that is influenced by a variety of external and internal variables, including environmental, demographic, and biopsychosocial factors, to determine the development and progression of age-related diseases, rather than being a solely static intrinsic process of cellular alterations.
1. Introduction
The dramatic improvement in life expectancy over the past century led to an unprecedented demographic shift toward an aging population; the proportion of the population over 65 is higher than ever before. As the population boom of the 20th century ages, age-related diseases have come to the forefront as emergent health concerns [1]. In contrast to maternal, infectious diseases that were widely prevalent and a primary health concern of the early 20th century, age-related diseases are often chronic and require continual treatment over an extended period of time, thus correlating increased lifespan with chronic disease onset and elevated expense burden. Aging is a multifactorial dynamic process that is influenced by a variety of external and internal variables, including environmental, demographic, and biopsychosocial factors, to determine the development and progression of age-related diseases, rather than being a solely static intrinsic process of cellular alterations (Figure 1).
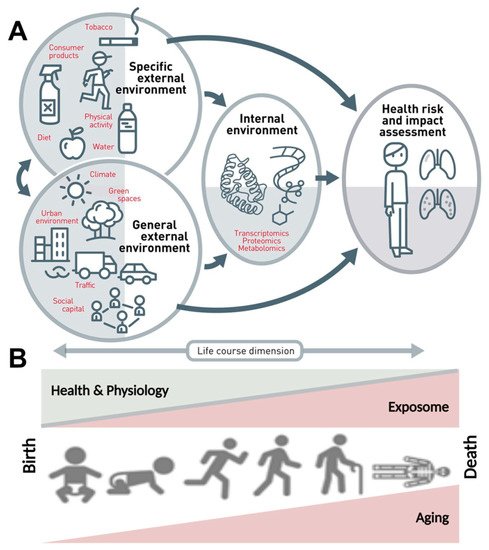
Figure 1. The exposome concept. (A) The exposome comprises the totality of a person’s external and internal exposures, from birth to death. (B) The external exposures and their internal exposure-related biochemical changes accumulate steadily over the aging process and lead to altered health risks. Adapted from Vrijheid et al. [2] (upper part, Copyright © 2021, BMJ Publishing Group Ltd. and the British Thoracic Society) and Misra [3] (lower part, under the terms of the Creative Commons Attribution License (CC BY), Copyright © 2021 Misra) with permission.
The aging population is particularly susceptible to cardiovascular disease (CVD), demonstrating the leading cause of death in populations aged over 65 years (Figure 2), and creating an urgent need for research in the field. Compounding the rise in CVD prevalence, as age advances, there is also a rise in complications and comorbidities of CVD [4][5][6]. This phenomenon is partly due to the “silent” nature of CVD pathophysiological development, but also due to vascular aging, which represents all changes in the vessels over time that exacerbate disease development [7]. Specifically, aged vessels have an impaired endothelium and other constitutional changes, which make them more prone to atherosclerotic lesions, vascular injury, and calcification, alongside blunted angiogenesis [8]. With age, the endothelium displays decreased responsiveness that manifests endothelial dysfunction in elderly people [9][10] and corresponds well with the demographic data that associates age, CVD incidence, and CVD comorbidities [11]. Endothelial impairment leading to endothelial dysfunction is also accompanied by smooth muscle changes, since arterial stiffening is also observed in aged vessels. Both mechanistic aspects correlate with future cardiovascular events in humans [12].
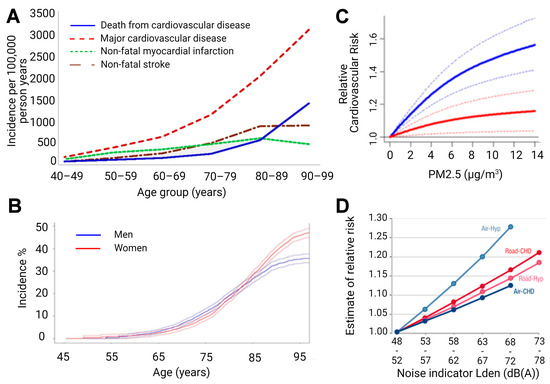
Figure 2. (A) Age-specific crude incidence of confirmed major cardiovascular disease by type of first event (non-fatal myocardial infarction, non-fatal stroke, and death from cardiovascular disease). Reused from [13] with permission under the terms of the Creative Commons Attribution Non-commercial License, Copyright © Driver et al. 2008. (B) Risk of common neurological diseases for 45-year-old men and women. In this analysis, follow-up ended at time of first occurrence of dementia, stroke, or parkinsonism. For instance, for individuals who first suffered a stroke and subsequently developed dementia, only the stroke event is considered. Reused from [14] with permission, Copyright © 2021, BMJ Publishing Group Ltd. All rights reserved. (C) Predicted values of relative risk for cardiovascular mortality by chronic exposure to increasing particulate matter concentrations for high ozone levels (37.60 ppb, solid blue line) and low ozone levels (20.26 ppb, solid red line) with uncertainty intervals (dashed lines). Reused from [15] with permission under the terms of the Creative Commons CC BY license, Copyright © 2021, The Author(s). (D) Exposure-response relationships for the associations between transportation noise and cardiovascular health outcomes. Road—road traffic noise, Air—aircraft noise, Hyp—hypertension, CHD—coronary heart disease, and Lden—day-evening-night level, i.e., the average sound pressure level measured over a 24-h period. Reused from [16] with permission, Copyright © 2021, Oxford University Press.
The same holds true for the aging brain (Figure 2). The incidence of stroke shows a dramatic increase in the elderly [17], and cognitive impairment clearly progresses with age and represents an accepted early diagnostic parameter for later dementia and neurodegeneration [18]. When looking at the risk factors of dementia in detail, it becomes clear that there is a large overlap with cardiovascular risk factors [19]. Mounting evidence indicates that the aging process is fundamentally driven by environmental exposures, and interestingly, age-related pathomechanisms were also observed in the context of predominant environmental pollutants, such as air pollution [20][21] and (traffic) noise exposure [22][23], with growing evidence suggesting that these pollutants might cause or accelerate age-related diseases.
2. Impact of Aging on Inflammation, Adverse Redox Signaling, Endothelial Dysfunction, and CVD
Endothelial dysfunction is an important indicator of subclinical CVD and serves as an early predictor of developing atherosclerosis, hypertension, and future cardiovascular events. There are two critical mechanisms through which endothelial dysfunction influences pathogenesis within the context of vascular aging. First, it promotes vasoconstriction, thrombocyte activation, leukocyte infiltration, and smooth muscle cell proliferation in the vessel wall; all of which precede cardiovascular events. The second is due to impaired endothelial signaling in all vessels; age-dependent endothelial dysfunction is found in both macrovessels and resistance vessels (for review see [24]), and thereby can impact a wide variety of disease states.
Three interdependent players are known to trigger endothelial dysfunction—inflammation, oxidative stress, and impaired nitric oxide (•NO) signaling [25]. Endothelial oxidative stress, an important trigger of endothelial dysfunction, is associated with age-related diseases other than CVD, including erectile dysfunction, renal dysfunction, Alzheimer’s disease, or retinopathy [26][27][28][29]. The studies of Mayhan et al. highlight these findings, demonstrating that cerebral arterioles show diminished eNOS-dependent reactivity, which positively correlated with increased oxidative stress in aged rats [30]. These findings were echoed in studies in other vessels and conditions, showing that endothelial dysfunction and oxidative stress are present in aging retinal vessels [31], and are a contributing factor in Alzheimer’s and Parkinson’s diseases, through several mechanisms [32][33]. Lastly, oxidative stress in combination with vascular inflammation and impaired •NO signaling were identified as key players in age-related endothelial dysfunction by our group and many others (for review see [34][35]). As aged vessels show strong associations with oxidative imbalances, inflammatory increases, and negative effects on •NO signaling, aging is implicated as an independent risk factor for CVD [36][37].
In many ways, a reciprocal and interdependent relationship exists between oxidative stress deriving from mitochondrial or enzymatic sources, endothelial dysfunction, and hypertension, diabetes, and atherosclerosis. It is unsurprising then, that oxidative stress [38][39], endothelial dysfunction [36], and the aforementioned CVDs [40][41] all see an increased incidence with advancing age, as they often occur in parallel, exert some influence on each other, and also have associations with low-level inflammation. Accordingly, age-dependent changes in the composition and function of high-density lipoproteins (HDL) were reported [42], which further underlined the contributing mechanisms of the risk factors previously discussed, since HDL inhibit inflammation, have antioxidant properties [43], and inversely correlate with coronary disease risk [43]. The degradation of HDL quality over time negatively impacts endothelial function, a critical factor in the initiation and development of atherosclerosis, potentially indicating HDL as a target for therapeutic intervention of age-related CVD [44].
Hypertension, the predominant risk factor for atherosclerosis and other CVD, potentiates the causative elements behind endothelial dysfunction, making effective treatment of hypertension an important route for the prevention of age-related CVD. To this end, pregnant spontaneously hypertensive rats were treated with nitrovasodilator pentaerythrityl tetranitrate, which demonstrated blood pressure lowering effects that were inherited by offspring. It was found that enhanced histone 3 lysine 27 acetylation and histone 3 lysine 4 trimethylation (epigenetic markers usually associated with transcriptional activation) promoted the transcriptional activation of cardioprotective genes like eNOS, SOD2, GPx-1, and HO-1, which explained the observed heritable effects [45]. Drugs with epigenetic effects, like pentaerithrityl tetranitrate, could conceivably be used to extend the number of healthy years, and perhaps stave off the effects of cardiovascular aging. A third possible therapeutic strategy would utilize mitochondria-targeted antioxidants to mitigate the “side effects” of the aging process. Treatment with dietary vitamins equating to unspecific antioxidant treatment was not found to be effective in preventing vascular aging. However, specifically targeting mitochondrial ROS could represent a possible strategy to alleviate, at least in part, age-related endothelial dysfunction. Along this line, age-related endothelial dysfunction was alleviated by administration of mito-quinone in mice [46]. Some risk factors for CVD could be changed by lifestyle alterations, such as smoking and diet [47], but aging is a factor that is not preventable, and so must be tackled in a bottom-up approach.
As previously mentioned, low-level inflammation is commonly found in aged individuals. One study found that plasma levels of important inflammatory markers, including soluble vascular adhesion molecule 1(sVCAM-1), interleukin 6 (IL-6), and monocyte chemoattractant protein 1 (MCP-1) positively correlated with age, even where there was no underlying CVD or risk factors present [48]. Another study found positive correlations between age and levels of circulating IL-6, IL-1 receptor antagonist (IL-1ra), IL-18, C-reactive protein (CRP), and fibrinogen, in both men and women, most persisting after correction for other risk factors. Increases of soluble IL-6 receptor (sIL-6r) occurred with greater age, but this effect was only noted in men [49]. A meta-analysis spanning 32 cross-sectional studies and over 23,000 subjects revealed associations between serum CRP and IL-6 levels and the onset or presence of frailty and pre-frailty, a phenotype that encompassed unintentional weight loss, exhaustion, weakness, slow walking speed, or low physical capability [50]. The hazard ratio for serum CRP levels and incidence of frailty was 1.06 (95% confidence interval [CI] 0.78–1.44), alongside a hazard ratio of 1.19 (95% CI 0.87–1.62) for IL-6, after adjustment for 9 potential confounders [50], illustrating a correlation between the presence of inflammation and age-related ailments.
Air and noise pollution are novel cardiovascular risk factors whose mechanisms are still being investigated, but have so far shown similar molecular signatures [51][52][53] to one another, as well as to classical risk factors like hypertension, hypercholesterolemia, or hyperglycemia [54][55][56]. Foremost amongst these signatures appears to be oxidative stress and inflammation, which both mediate the detrimental effects following exposure to noise and air pollution [57]. Despite some clarity as to the mechanisms, it is not fully understood how crosstalk between stress response pathways, redox signaling, and the immune system coordinate to cause cardiovascular damage in response to these novel risk factors.
3. Impact of Aging on Inflammation, Adverse Redox Signaling, Neuronal Degeneration, and Neurological Disease
Since CVDs and neurological diseases have a substantial overlap in risk factors and pathophysiological pathways, it is important to highlight the aforementioned mechanisms of action in the context of aging and neurological disease. In general, functional and structural deterioration of the aging brain is a cumulative process that starts with subclinical alterations at the molecular level. These changes include accumulation of mutations, telomere attrition, and epigenetic alterations, resulting in genomic instability and thus priming for neuronal damage and loss, reduced neurotransmitter levels, enhanced neuroinflammation, increased susceptibility to cerebral ROS, and decreased cerebral vascular compliance. All of these adverse processes are associated with increased risk of age-related neurological diseases, such as stroke, epilepsy, Parkinson’s disease, and dementia/cognitive decline [58]. Immunosenescence and inflammaging, as the most recognized effects of aging [59], might promote neuroinflammatory processes along with cerebral oxidative stress, via altered microglia activation (immune cells of the brain), which are central to neurotoxicity through the release of neurotoxic cytokines, such as TNFα, IL-1β, and INF-γ, as well as different ROS such as ONOO− and O2•− [60][61]. Microglial dysregulation represents a hallmark of various neurological complications, and adverse redox regulation of and by microglia plays a crucial role in these processes [62][63][64]. Neuroinflammation and cerebral oxidative stress might act together to increase neuronal damage/loss and amyloid deposition, as well as to decrease cerebral •NO bioavailability via NOX-2 activation and uncoupling of neuronal •NO synthase (nNOS), leading to cerebral vascular endothelial dysfunction and ultimately contributing to increased risk of stroke, epilepsy, Parkinson’s disease, and dementia/cognitive decline in the elderly [21].
From an epidemiological point of view, the accelerated aging of the population and the correspondent increase in the elderly would affect the number of patients with neurological diseases, as recently demonstrated by results of the Dijon Stroke Registry. In this study, an increase of 55% in the total annual number of stroke cases by 2030 was calculated, largely driven by increased prevalence in the group of elderly people (65% in people ≥ 75 years vs. 25% in people < 75 years) [65]. Importantly, data from the Framingham study demonstrated older age at stroke onset, but not gender or stroke type, to be associated with increased disability [66]. Further epidemiological studies revealed a strong age-dependency for the incidence of epilepsy [67], Parkinson’s disease [68], and dementia [69], with a continuous and strong growth in numbers in the elderly. Thus, the coincidence of CVDs and neurological diseases in the elderly is not surprising, due to shared risk factors, which themselves express a high age-dependency, such as hypertension, diabetes, vascular dysfunction, and atherosclerosis, accompanied by altered molecular mechanisms centered on inflammation and adverse redox signaling.
4. The Oxidative Stress Concept of Aging
In 1954, Harman first described the “free radical theory of aging” [70]. He reasoned that since aging is a universal phenomenon, the underlying causation must also be universally present in all organisms. To this end, the focus shifted toward the hydroxyl radical and molecular oxygen being important mediators of the aging process [71]. Mitochondria are prolific producers of ROS within the cell, so they were natural targets for investigation within this theory. Since this high concentration of mitochondrial ROS (mtROS) is likely partly responsible for the high mutation rate of mtDNA, it is therefore necessary that two spatially separated genomes (nuclear and mitochondrial) co-exist, and both are required for the assembly of the respiratory chain components [72]. Further, as the mitochondrial genome malfunctions, irregularities in physiology and ATP synthesis are also seen, which are accompanied by amplified ROS generation and increased apoptosis [73]. Within the context of aging, the focus shifted away specifically from the hydroxyl radical and onto another free radical species, •NO, which is now known to be an important vasodilator, to play a role in vascular smooth muscle cell proliferation, and to inhibit platelet aggregation, amongst other important regulatory roles. The age-dependent impairment of vascular redox balance is strongly linked to the bioavailability of the •NO radical [74], which could be reduced through consumption by superoxide, and consequently lead to impaired vasorelaxation [36][75]. •NO could thereby potentially serve as a biomarker for age-dependent endothelial dysfunction.
The free radical theory of aging was amended in 1972 by Harman to delineate the specific role of mitochondria [76], which were then moved to the forefront of the field. Harmon proposed that the mitochondrion was the primary origin of oxidative stress as well as the target—mitochondria produce a significant amount of cellular energy but are also damaged by ROS, which can attack both mitochondrial and nuclear DNA and can cause significant damage. With age, the damage accrued can result in defective mitochondria, which produce more and more ROS and in turn cause more oxidant-induced mutations and deletions, and culminate in a loss of cellular function, apoptosis, and necrosis. To this end, oxidant-induced damage in mtDNA was reported in the form of 8-oxo-deoxyguanosine (8-oxo-dG) [77][78], a mutagenic lesion whose accumulation was linked to pathological processes [79], and inversely correlated with lifespan of short-lived animals in the nuclear DNA and mtDNA of cardiac tissue. In brain tissue of long-lived animals, however, 8-oxo-dG content was higher in nuclear DNA (data not shown) [80]. These insights could be partially explained by higher metabolic rate, lower antioxidant clearance and defense, and possibly less efficient DNA repair. In this manner, genomic instability and cellular senescence occur as a result of age-related oxidative stress-induced DNA damages associated with shortened telomeres, increased DNA methylation, and decreased DNA content, all of which contribute to numerous degenerative and aging-related diseases [81]. In two mouse knockout models (ALDH-2−/−, MnSOD−/−), we found that mitochondrial ROS, mitochondrial DNA (mtDNA), and vascular dysfunction positively correlated with age [82]. Further, our data showed a correlation between endothelial dysfunction and mitochondrial ROS, which itself was mostly dependent on age, but secondarily dependent on the level of antioxidant enzymes present. Our data also showed a correlation between mtROS and mtDNA strand breaks, which led to a reasoning that mtDNA strand breaks arise from mtROS through direct interaction and oxidative DNA lesions, and given enough time and stress, could result in mitochondrial uncoupling and a secondary increase in ROS generation (through impaired de novo synthesis of functional respiratory complexes, due to mtDNA degradation or mutation). The ultimate message of the free radical hypothesis of aging is that ROS cause substantive alterations in biological macromolecules over the organism’s lifespan, which accumulate to detrimental effect [83]. The conclusion can be made that accumulation of DNA damage in sum cannot be a “one size fits all” predictor of total life years, but that the kinetics of formation and repair of DNA damage will vary, depending on species and tissue.
However, ROS generation is not the sole factor in the slow degradation of vascular function. Antioxidant defense and clearance are also impacted by age. For example, cytosolic superoxide dismutase 1 (SOD1) [75][84], mitochondrial superoxide dismutase 2 (SOD2) [85], extracellular SOD (ecSOD), and thioredoxin-1 (Trx) [86] showed both age- and expression-related reductions in clearance efficacy, as reflected by studies of endothelial function in young and old mice. If superoxide is the major contributor to vascular aging, the question arises—why are these antioxidant systems seemingly unable to defend against increasing levels of oxidative stress? To that point, in aging vessels, SOD2 was found to be heavily nitrated, and its activity thereby impaired. These findings were accompanied by increased 3-nitrotyrosine staining, which implies a role for peroxynitrite, a product of superoxide and •NO as the nitrating agent [87]. It is obvious then that oxidative burden can cause inhibition of this protective enzyme, which then perpetuates a vicious cycle leaving the enzyme unable to perform effectively. Though it would be intuitive to expect that the overexpression of antioxidant enzymes would result in expansion of lifespan, this was not shown (SOD2+/− or SOD2tg, GPx-1−/−, GPx-4−/− or MsrA−/−, SOD1tg, catalasetg) [88], though overexpression of Trx1 was shown to increase lifespan and stress resistance [89]. Conversely, only SOD1−/− mice and mice with double gene ablation combinations reduced life expectancy [88][90], and SOD2 knockouts do not survive past a few weeks from birth [91][92]. Taken in conjunction, these data suggest that antioxidant systems are critically important to life, but also that there is a “cap” to their beneficial effects. This further implies that it is not the absolute amount of oxidative stress that impacts lifespan, but rather, a balance that must be maintained. While oxidative stress might not be the direct determinant in aging, as previously hypothesized [88][90][93], the contribution of oxidative stress in aging seems to be a factor that prevents healthy aging by impacting organ function [94][95][96][97].
The length of time an organism remains healthy is another factor through which antioxidant enzymes could have a significant effect and a notable clinical importance. This “healthspan” could be indicated by the lack or decreased progression of age-related cardiovascular complications and the resistance to stress conditions during normal aging [89]. In studies utilizing genetic deletion of aldehyde dehydrogenase-2 (ALDH-2) and manganese superoxide dismutase (MnSOD), two important mitochondrial antioxidant proteins, we found that mitochondrial oxidative stress and vascular dysfunction arose as a function of aging [82], which supports the idea that mtROS is especially important in the degree of health in aging [94][95][96][97].
Aside from mitochondrial ROS, there are other cellular sources of ROS that have an impact on the healthspan. The state of eNOS plays an important role in whether it produces a vascular hero, •NO, or a villain, O2•− [98]. In the coupled state, eNOS consists of a protein dimer and two BH4 cofactors that facilitate electron transfer needed for L-arginine oxidation and production of •NO [54][55][56]. When BH4 is either oxidized to BH2 or absent, or electron flow from the reductase to the oxygenase domain is impaired by either eNOS S-glutathionylation or adverse phosphorylation, the eNOS dimer is uncoupled and produces ROS in the form of O2•− (which is why NOS enzymes are also called Janus-faced enzymes). The overproduction of O2•− further oxidizes BH4 and inhibits •NO synthesis. The result of eNOS uncoupling is then a reduction in •NO bioavailability [99] and can contribute to the pathogenesis of endothelial dysfunction in aged vessels [9][36]. It was reported from several sources that eNOS expression levels rise with age, which could possibly be to counteract the effect of eNOS uncoupling and reduced •NO bioavailability. There are also groups who report unchanged eNOS expression in aged vessels, but instead report decreases in Akt-dependent phosphorylation of eNOSSer1177. Either reports could be consistent with the findings of endothelial dysfunction in aged vessels and elderly individuals [100]. We additionally reported both S-glutathionylation by PKC and adverse phosphorylation of eNOS at Thr495 and Tyr657 by PYK-2, as important redox-sensitive mechanisms in the process of aging-induced vascular dysfunction [101].
References
- Kelly, D.T. Paul dudley white international lecture. Our future society. A global challenge. Circulation 1997, 95, 2459–2464.
- Vrijheid, M. The exposome: A new paradigm to study the impact of environment on health. Thorax 2014, 69, 876–878.
- Misra, B.B. The chemical exposome of human aging. Front. Genet. 2020, 11, 574936.
- Cesari, M.; Onder, G.; Russo, A.; Zamboni, V.; Barillaro, C.; Ferrucci, L.; Pahor, M.; Bernabei, R.; Landi, F. Comorbidity and physical function: Results from the aging and longevity study in the sirente geographic area (ilsirente study). Gerontology 2006, 52, 24–32.
- Yancik, R.; Ershler, W.; Satariano, W.; Hazzard, W.; Cohen, H.J.; Ferrucci, L. Report of the national institute on aging task force on comorbidity. J. Gerontol. Ser. A Biol. Sci. Med. Sci. 2007, 62, 275–280.
- Wieland, G.D. From bedside to bench: Research in comorbidity and aging. Sci. Aging Knowl. Environ. Sage Ke 2005, 2005, pe29.
- Lakatta, E.G.; Levy, D. Arterial and cardiac aging: Major shareholders in cardiovascular disease enterprises: Part i: Aging arteries: A “set up” for vascular disease. Circulation 2003, 107, 139–146.
- Herrera, M.D.; Mingorance, C.; Rodriguez-Rodriguez, R.; Alvarez de Sotomayor, M. Endothelial dysfunction and aging: An update. Ageing Res. Rev. 2010, 9, 142–152.
- Seals, D.R.; Jablonski, K.L.; Donato, A.J. Aging and vascular endothelial function in humans. Clin. Sci. (Lond.) 2011, 120, 357–375.
- Tanaka, H.; Dinenno, F.A.; Seals, D.R. Age-related increase in femoral intima-media thickness in healthy humans. Arter. Thromb. Vasc. Biol. 2000, 20, 2172.
- Bischoff, B.; Silber, S.; Richartz, B.M.; Pieper, L.; Klotsche, J.; Wittchen, H.U. Inadequate medical treatment of patients with coronary artery disease by primary care physicians in germany. Clin. Res. Cardiol. 2006, 95, 405–412.
- Ras, R.T.; Streppel, M.T.; Draijer, R.; Zock, P.L. Flow-mediated dilation and cardiovascular risk prediction: A systematic review with meta-analysis. Int. J. Cardiol. 2013, 168, 344–351.
- Driver, J.A.; Djousse, L.; Logroscino, G.; Gaziano, J.M.; Kurth, T. Incidence of cardiovascular disease and cancer in advanced age: Prospective cohort study. BMJ 2008, 337, a2467.
- Licher, S.; Darweesh, S.K.L.; Wolters, F.J.; Fani, L.; Heshmatollah, A.; Mutlu, U.; Koudstaal, P.J.; Heeringa, J.; Leening, M.J.G.; Ikram, M.K.; et al. Lifetime risk of common neurological diseases in the elderly population. J. Neurol. Neurosurg. Psychiatry 2019, 90, 148–156.
- Weichenthal, S.; Pinault, L.L.; Burnett, R.T. Impact of oxidant gases on the relationship between outdoor fine particulate air pollution and nonaccidental, cardiovascular, and respiratory mortality. Sci. Rep. 2017, 7, 16401.
- Munzel, T.; Sorensen, M.; Gori, T.; Schmidt, F.P.; Rao, X.; Brook, J.; Chen, L.C.; Brook, R.D.; Rajagopalan, S. Environmental stressors and cardio-metabolic disease: Part i-epidemiologic evidence supporting a role for noise and air pollution and effects of mitigation strategies. Eur. Heart J. 2017, 38, 550–556.
- Kelly-Hayes, M. Influence of age and health behaviors on stroke risk: Lessons from longitudinal studies. J. Am. Geriatr. Soc. 2010, 58 (Suppl. 2), S325–S328.
- Gauthier, S.; Reisberg, B.; Zaudig, M.; Petersen, R.C.; Ritchie, K.; Broich, K.; Belleville, S.; Brodaty, H.; Bennett, D.; Chertkow, H.; et al. Mild cognitive impairment. Lancet 2006, 367, 1262–1270.
- Barnes, D.E.; Yaffe, K. The projected effect of risk factor reduction on alzheimer’s disease prevalence. Lancet Neurol. 2011, 10, 819–828.
- Daiber, A.; Kuntic, M.; Hahad, O.; Delogu, L.G.; Rohrbach, S.; Di Lisa, F.; Schulz, R.; Munzel, T. Effects of air pollution particles (ultrafine and fine particulate matter) on mitochondrial function and oxidative stress—implications for cardiovascular and neurodegenerative diseases. Arch Biochem. Biophys. 2020, 696, 108662.
- Hahad, O.; Lelieveld, J.; Birklein, F.; Lieb, K.; Daiber, A.; Munzel, T. Ambient air pollution increases the risk of cerebrovascular and neuropsychiatric disorders through induction of inflammation and oxidative stress. Int. J. Mol. Sci. 2020, 21, 4306.
- Daiber, A.; Kroller-Schon, S.; Oelze, M.; Hahad, O.; Li, H.; Schulz, R.; Steven, S.; Munzel, T. Oxidative stress and inflammation contribute to traffic noise-induced vascular and cerebral dysfunction via uncoupling of nitric oxide synthases. Redox Biol. 2020, 34, 101506.
- Hahad, O.; Prochaska, J.H.; Daiber, A.; Muenzel, T. Environmental noise-induced effects on stress hormones, oxidative stress, and vascular dysfunction: Key factors in the relationship between cerebrocardiovascular and psychological disorders. Oxidative Med. Cell. Longev. 2019, 2019, 4623109.
- Crimi, E.; Ignarro, L.J.; Napoli, C. Microcirculation and oxidative stress. Free Radic. Res. 2007, 41, 1364–1375.
- El Assar, M.; Angulo, J.; Rodriguez-Manas, L. Oxidative stress and vascular inflammation in aging. Free Radic. Biol. Med. 2013, 65, 380–401.
- Burnett, A.L. The role of nitric oxide in erectile dysfunction: Implications for medical therapy. J. Clin. Hypertens. 2006, 8, 53–62.
- Csiszar, A.; Toth, J.; Peti-Peterdi, J.; Ungvari, Z. The aging kidney: Role of endothelial oxidative stress and inflammation. Acta Physiol. Hung. 2007, 94, 107–115.
- Price, J.M.; Hellermann, A.; Hellermann, G.; Sutton, E.T. Aging enhances vascular dysfunction induced by the alzheimer’s peptide beta-amyloid. Neurol. Res. 2004, 26, 305–311.
- Coleman, H.R.; Chan, C.C.; Ferris, F.L., 3rd; Chew, E.Y. Age-related macular degeneration. Lancet 2008, 372, 1835–1845.
- Mayhan, W.G.; Arrick, D.M.; Sharpe, G.M.; Sun, H. Age-related alterations in reactivity of cerebral arterioles: Role of oxidative stress. Microcirculation 2008, 15, 225–236.
- Militante, J.; Lombardini, J.B. Age-related retinal degeneration in animal models of aging: Possible involvement of taurine deficiency and oxidative stress. Neurochem. Res. 2004, 29, 151–160.
- Fischer, R.; Maier, O. Interrelation of oxidative stress and inflammation in neurodegenerative disease: Role of tnf. Oxidative Med. Cell. Longev. 2015, 2015, 610813.
- Blasiak, J.; Petrovski, G.; Vereb, Z.; Facsko, A.; Kaarniranta, K. Oxidative stress, hypoxia, and autophagy in the neovascular processes of age-related macular degeneration. BioMed Res. Int. 2014, 2014, 768026.
- Daiber, A.; Kienhoefer, J.; Zee, R.; Ullrich, V.; Van der Loo, B.; Bachschmid, M. The role of mitochondrial reactive oxygen species formation for age-induced vascular dysfunction. In Aging and Age-Related Disorders; Bondy, S.C., Maiese, K., Eds.; Springer (Humana Press): Totowa, NJ, USA, 2010; pp. 237–257.
- Mikhed, Y.; Daiber, A.; Steven, S. Mitochondrial oxidative stress, mitochondrial DNA damage and their role in age-related vascular dysfunction. Int. J. Mol. Sci. 2015, 16, 15918–15953.
- Gerhard, M.; Roddy, M.A.; Creager, S.J.; Creager, M.A. Aging progressively impairs endothelium-dependent vasodilation in forearm resistance vessels of humans. Hypertension 1996, 27, 849–853.
- Jousilahti, P.; Vartiainen, E.; Tuomilehto, J.; Puska, P. Sex, age, cardiovascular risk factors, and coronary heart disease: A prospective follow-up study of 14 786 middle-aged men and women in finland. Circulation 1999, 99, 1165–1172.
- Kimura, Y.; Matsumoto, M.; Den, Y.B.; Iwai, K.; Munehira, J.; Hattori, H.; Hoshino, T.; Yamada, K.; Kawanishi, K.; Tsuchiya, H. Impaired endothelial function in hypertensive elderly patients evaluated by high resolution ultrasonography. Can. J. Cardiol. 1999, 15, 563–568.
- Wray, D.W.; Nishiyama, S.K.; Harris, R.A.; Zhao, J.; McDaniel, J.; Fjeldstad, A.S.; Witman, M.A.; Ives, S.J.; Barrett-O’Keefe, Z.; Richardson, R.S. Acute reversal of endothelial dysfunction in the elderly after antioxidant consumption. Hypertension 2012, 59, 818–824.
- Savji, N.; Rockman, C.B.; Skolnick, A.H.; Guo, Y.; Adelman, M.A.; Riles, T.; Berger, J.S. Association between advanced age and vascular disease in different arterial territories: A population database of over 3.6 million subjects. J. Am. Coll Cardiol. 2013, 61, 1736–1743.
- Ong, K.L.; Cheung, B.M.; Man, Y.B.; Lau, C.P.; Lam, K.S. Prevalence, awareness, treatment, and control of hypertension among united states adults 1999–2004. Hypertension 2007, 49, 69–75.
- Holzer, M.; Trieb, M.; Konya, V.; Wadsack, C.; Heinemann, A.; Marsche, G. Aging affects high-density lipoprotein composition and function. Biochim. Biophys. Acta 2013, 1831, 1442–1448.
- Tabet, F.; Rye, K.A. High-density lipoproteins, inflammation and oxidative stress. Clin. Sci. (Lond.) 2009, 116, 87–98.
- Besler, C.; Heinrich, K.; Riwanto, M.; Luscher, T.F.; Landmesser, U. High-density lipoprotein-mediated anti-atherosclerotic and endothelial-protective effects: A potential novel therapeutic target in cardiovascular disease. Curr. Pharm. Des. 2010, 16, 1480–1493.
- Wu, Z.; Siuda, D.; Xia, N.; Reifenberg, G.; Daiber, A.; Munzel, T.; Forstermann, U.; Li, H. Maternal treatment of spontaneously hypertensive rats with pentaerythritol tetranitrate reduces blood pressure in female offspring. Hypertension 2015, 65, 232–237.
- Gioscia-Ryan, R.A.; LaRocca, T.J.; Sindler, A.L.; Zigler, M.C.; Murphy, M.P.; Seals, D.R. Mitochondria-targeted antioxidant (mitoq) ameliorates age-related arterial endothelial dysfunction in mice. J. Physiol. 2014, 592, 2549–2561.
- Klipstein-Grobusch, K.; Geleijnse, J.M.; den Breeijen, J.H.; Boeing, H.; Hofman, A.; Grobbee, D.E.; Witteman, J.C. Dietary antioxidants and risk of myocardial infarction in the elderly: The rotterdam study. Am. J. Clin. Nutr. 1999, 69, 261–266.
- Miles, E.A.; Rees, D.; Banerjee, T.; Cazzola, R.; Lewis, S.; Wood, R.; Oates, R.; Tallant, A.; Cestaro, B.; Yaqoob, P.; et al. Age-related increases in circulating inflammatory markers in men are independent of bmi, blood pressure and blood lipid concentrations. Atherosclerosis 2008, 196, 298–305.
- Ferrucci, L.; Corsi, A.; Lauretani, F.; Bandinelli, S.; Bartali, B.; Taub, D.D.; Guralnik, J.M.; Longo, D.L. The origins of age-related proinflammatory state. Blood 2005, 105, 2294–2299.
- Soysal, P.; Stubbs, B.; Lucato, P.; Luchini, C.; Solmi, M.; Peluso, R.; Sergi, G.; Isik, A.T.; Manzato, E.; Maggi, S.; et al. Inflammation and frailty in the elderly: A systematic review and meta-analysis. Ageing Res. Rev. 2016, 31, 1–8.
- Miguel, V.; Cui, J.Y.; Daimiel, L.; Espinosa-Diez, C.; Fernandez-Hernando, C.; Kavanagh, T.J.; Lamas, S. The role of micrornas in environmental risk factors, noise-induced hearing loss, and mental stress. Antioxid. Redox Signal. 2018, 28, 773–796.
- Golbidi, S.; Li, H.; Laher, I. Oxidative stress: A unifying mechanism for cell damage induced by noise, (water-pipe) smoking, and emotional stress-therapeutic strategies targeting redox imbalance. Antioxid. Redox Signal. 2018, 28, 741–759.
- Ghezzi, P.; Floridi, L.; Boraschi, D.; Cuadrado, A.; Manda, G.; Levic, S.; D’Acquisto, F.; Hamilton, A.; Athersuch, T.J.; Selley, L. Oxidative stress and inflammation induced by environmental and psychological stressors: A biomarker perspective. Antioxid. Redox Signal. 2018, 28, 852–872.
- Daiber, A.; Steven, S.; Weber, A.; Shuvaev, V.V.; Muzykantov, V.R.; Laher, I.; Li, H.; Lamas, S.; Munzel, T. Targeting vascular (endothelial) dysfunction. Br. J. Pharm. 2017, 174, 1591–1619.
- Daiber, A.; Di Lisa, F.; Oelze, M.; Kroller-Schon, S.; Steven, S.; Schulz, E.; Munzel, T. Crosstalk of mitochondria with nadph oxidase via reactive oxygen and nitrogen species signalling and its role for vascular function. Br. J. Pharm. 2017, 174, 1670–1689.
- Wenzel, P.; Kossmann, S.; Munzel, T.; Daiber, A. Redox regulation of cardiovascular inflammation—Immunomodulatory function of mitochondrial and nox-derived reactive oxygen and nitrogen species. Free Radic. Biol. Med. 2017, 109, 48–60.
- Munzel, T.; Daiber, A. Environmental stressors and their impact on health and disease with focus on oxidative stress. Antioxid. Redox Signal. 2018, 28, 735–740.
- Kowalska, M.; Owecki, M.; Prendecki, M.; Wize, K.; Nowakowska, J.; Kozubski, W.; Lianeri, M.; Dorszewska, J. Aging and neurological diseases. In Senescence—Physiology or Pathology; Kozubski, W., Dorszewska, J., Eds.; IntechOpen: London, UK, 2017.
- Franceschi, C.; Capri, M.; Monti, D.; Giunta, S.; Olivieri, F.; Sevini, F.; Panourgia, M.P.; Invidia, L.; Celani, L.; Scurti, M.; et al. Inflammaging and anti-inflammaging: A systemic perspective on aging and longevity emerged from studies in humans. Mech. Ageing Dev. 2007, 128, 92–105.
- Brown, G.C.; Vilalta, A. How microglia kill neurons. Brain Res. 2015, 1628, 288–297.
- Block, M.L.; Zecca, L.; Hong, J.S. Microglia-mediated neurotoxicity: Uncovering the molecular mechanisms. Nat. Rev. Neurosci. 2007, 8, 57–69.
- Haslund-Vinding, J.; McBean, G.; Jaquet, V.; Vilhardt, F. Nadph oxidases in oxidant production by microglia: Activating receptors, pharmacology and association with disease. Br. J. Pharm. 2017, 174, 1733–1749.
- McBean, G.J.; Lopez, M.G.; Wallner, F.K. Redox-based therapeutics in neurodegenerative disease. Br. J. Pharm. 2017, 174, 1750–1770.
- Vilhardt, F.; Haslund-Vinding, J.; Jaquet, V.; McBean, G. Microglia antioxidant systems and redox signalling. Br. J. Pharm. 2017, 174, 1719–1732.
- Bejot, Y.; Bailly, H.; Graber, M.; Garnier, L.; Laville, A.; Dubourget, L.; Mielle, N.; Chevalier, C.; Durier, J.; Giroud, M. Impact of the ageing population on the burden of stroke: The dijon stroke registry. Neuroepidemiology 2019, 52, 78–85.
- Kelly-Hayes, M.; Beiser, A.; Kase, C.S.; Scaramucci, A.; D’Agostino, R.B.; Wolf, P.A. The influence of gender and age on disability following ischemic stroke: The framingham study. J. Stroke Cereb. Dis. 2003, 12, 119–126.
- Olafsson, E.; Ludvigsson, P.; Gudmundsson, G.; Hesdorffer, D.; Kjartansson, O.; Hauser, W.A. Incidence of unprovoked seizures and epilepsy in iceland and assessment of the epilepsy syndrome classification: A prospective study. Lancet Neurol. 2005, 4, 627–634.
- de Lau, L.M.; Giesbergen, P.C.; de Rijk, M.C.; Hofman, A.; Koudstaal, P.J.; Breteler, M.M. Incidence of parkinsonism and parkinson disease in a general population: The rotterdam study. Neurology 2004, 63, 1240–1244.
- Corrada, M.M.; Brookmeyer, R.; Paganini-Hill, A.; Berlau, D.; Kawas, C.H. Dementia incidence continues to increase with age in the oldest old: The 90+ study. Ann. Neurol. 2010, 67, 114–121.
- Harman, D. Aging: A theory based on free radical and radiation chemistry. J. Gerontol. 1956, 11, 298–300.
- Waters, W.A. Some recent developments in the chemistry of free radicals. J. Chem. Soc. 1946, 409–415.
- Rogell, B.; Dean, R.; Lemos, B.; Dowling, D.K. Mito-nuclear interactions as drivers of gene movement on and off the x-chromosome. BMC Genom. 2014, 15, 330.
- Cadenas, E.; Davies, K.J. Mitochondrial free radical generation, oxidative stress, and aging. Free Radic. Biol. Med. 2000, 29, 222–230.
- Thomas, D.D.; Ridnour, L.A.; Isenberg, J.S.; Flores-Santana, W.; Switzer, C.H.; Donzelli, S.; Hussain, P.; Vecoli, C.; Paolocci, N.; Ambs, S.; et al. The chemical biology of nitric oxide: Implications in cellular signaling. Free Radic Biol. Med. 2008, 45, 18–31.
- van der Loo, B.; Bachschmid, M.; Skepper, J.N.; Labugger, R.; Schildknecht, S.; Hahn, R.; Mussig, E.; Gygi, D.; Luscher, T.F. Age-associated cellular relocation of sod 1 as a self-defense is a futile mechanism to prevent vascular aging. Biochem. Biophys. Res. Commun. 2006, 344, 972–980.
- Harman, D. The biologic clock: The mitochondria? J. Am. Geriatr. Soc. 1972, 20, 145–147.
- de Souza-Pinto, N.C.; Eide, L.; Hogue, B.A.; Thybo, T.; Stevnsner, T.; Seeberg, E.; Klungland, A.; Bohr, V.A. Repair of 8-oxodeoxyguanosine lesions in mitochondrial dna depends on the oxoguanine dna glycosylase (ogg1) gene and 8-oxoguanine accumulates in the mitochondrial dna of ogg1-defective mice. Cancer Res. 2001, 61, 5378–5381.
- de Souza-Pinto, N.C.; Hogue, B.A.; Bohr, V.A. DNA repair and aging in mouse liver: 8-oxodg glycosylase activity increase in mitochondrial but not in nuclear extracts. Free Radic. Biol. Med. 2001, 30, 916–923.
- Souza-Pinto, N.C.; Croteau, D.L.; Hudson, E.K.; Hansford, R.G.; Bohr, V.A. Age-associated increase in 8-oxo-deoxyguanosine glycosylase/ap lyase activity in rat mitochondria. Nucleic Acids Res. 1999, 27, 1935–1942.
- Barja, G.; Herrero, A. Oxidative damage to mitochondrial DNA is inversely related to maximum life span in the heart and brain of mammals. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2000, 14, 312–318.
- Barnes, R.P.; Fouquerel, E.; Opresko, P.L. The impact of oxidative DNA damage and stress on telomere homeostasis. Mech. Ageing Dev. 2019, 177, 37–45.
- Wenzel, P.; Schuhmacher, S.; Kienhofer, J.; Muller, J.; Hortmann, M.; Oelze, M.; Schulz, E.; Treiber, N.; Kawamoto, T.; Scharffetter-Kochanek, K.; et al. Manganese superoxide dismutase and aldehyde dehydrogenase deficiency increase mitochondrial oxidative stress and aggravate age-dependent vascular dysfunction. Cardiovasc Res. 2008, 80, 280–289.
- Kujoth, G.C.; Hiona, A.; Pugh, T.D.; Someya, S.; Panzer, K.; Wohlgemuth, S.E.; Hofer, T.; Seo, A.Y.; Sullivan, R.; Jobling, W.A.; et al. Mitochondrial DNA mutations, oxidative stress, and apoptosis in mammalian aging. Science 2005, 309, 481–484.
- Didion, S.P.; Kinzenbaw, D.A.; Schrader, L.I.; Faraci, F.M. Heterozygous cuzn superoxide dismutase deficiency produces a vascular phenotype with aging. Hypertension 2006, 48, 1072–1079.
- Brown, K.A.; Didion, S.P.; Andresen, J.J.; Faraci, F.M. Effect of aging, mnsod deficiency, and genetic background on endothelial function: Evidence for mnsod haploinsufficiency. Arter. Thromb. Vasc. Biol. 2007, 27, 1941–1946.
- Altschmied, J.; Haendeler, J. Thioredoxin-1 and endothelial cell aging: Role in cardiovascular diseases. Antioxid. Redox Signal. 2009, 11, 1733–1740.
- Goldstein, S.; Czapski, G.; Lind, J.; Merenyi, G. Tyrosine nitration by simultaneous generation of (.)no and o-(2) under physiological conditions. How the radicals do the job. J. Biol. Chem. 2000, 275, 3031–3036.
- Perez, V.I.; Bokov, A.; Van Remmen, H.; Mele, J.; Ran, Q.; Ikeno, Y.; Richardson, A. Is the oxidative stress theory of aging dead? Biochim. Biophys. Acta 2009, 1790, 1005–1014.
- Salmon, A.B.; Richardson, A.; Perez, V.I. Update on the oxidative stress theory of aging: Does oxidative stress play a role in aging or healthy aging? Free Radic. Biol. Med. 2010, 48, 642–655.
- Muller, F.L.; Lustgarten, M.S.; Jang, Y.; Richardson, A.; Van Remmen, H. Trends in oxidative aging theories. Free Radic. Biol. Med. 2007, 43, 477–503.
- Lebovitz, R.M.; Zhang, H.; Vogel, H.; Cartwright, J., Jr.; Dionne, L.; Lu, N.; Huang, S.; Matzuk, M.M. Neurodegeneration, myocardial injury, and perinatal death in mitochondrial superoxide dismutase-deficient mice. Proc. Natl. Acad. Sci. USA 1996, 93, 9782–9787.
- Li, Y.; Huang, T.T.; Carlson, E.J.; Melov, S.; Ursell, P.C.; Olson, J.L.; Noble, L.J.; Yoshimura, M.P.; Berger, C.; Chan, P.H.; et al. Dilated cardiomyopathy and neonatal lethality in mutant mice lacking manganese superoxide dismutase. Nat. Genet. 1995, 11, 376–381.
- Jang, Y.C.; Remmen, H.V. The mitochondrial theory of aging: Insight from transgenic and knockout mouse models. Exp. Gerontol. 2009, 44, 256–260.
- Dai, D.F.; Chiao, Y.A.; Marcinek, D.J.; Szeto, H.H.; Rabinovitch, P.S. Mitochondrial oxidative stress in aging and healthspan. Longev. Healthspan 2014, 3, 6.
- Hamilton, R.T.; Walsh, M.E.; Van Remmen, H. Mouse models of oxidative stress indicate a role for modulating healthy aging. J. Clin. Exp. Pathol. 2012, (Suppl. 4).
- Berry, A.; Cirulli, F. The p66(shc) gene paves the way for healthspan: Evolutionary and mechanistic perspectives. Neurosci. Biobehav. Rev. 2013, 37, 790–802.
- Wanagat, J.; Dai, D.F.; Rabinovitch, P. Mitochondrial oxidative stress and mammalian healthspan. Mech. Ageing Dev. 2010, 131, 527–535.
- Forstermann, U.; Munzel, T. Endothelial nitric oxide synthase in vascular disease: From marvel to menace. Circulation 2006, 113, 1708–1714.
- Schulz, E.; Jansen, T.; Wenzel, P.; Daiber, A.; Munzel, T. Nitric oxide, tetrahydrobiopterin, oxidative stress, and endothelial dysfunction in hypertension. Antioxid. Redox Signal 2008, 10, 1115–1126.
- Soucy, K.G.; Ryoo, S.; Benjo, A.; Lim, H.K.; Gupta, G.; Sohi, J.S.; Elser, J.; Aon, M.A.; Nyhan, D.; Shoukas, A.A.; et al. Impaired shear stress-induced nitric oxide production through decreased nos phosphorylation contributes to age-related vascular stiffness. J. Appl. Physiol. 2006, 101, 1751–1759.
- Oelze, M.; Kroller-Schon, S.; Steven, S.; Lubos, E.; Doppler, C.; Hausding, M.; Tobias, S.; Brochhausen, C.; Li, H.; Torzewski, M.; et al. Glutathione peroxidase-1 deficiency potentiates dysregulatory modifications of endothelial nitric oxide synthase and vascular dysfunction in aging. Hypertension 2014, 63, 390–396.

