 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Kun Song | + 3648 word(s) | 3648 | 2021-04-01 15:10:48 | | | |
| 2 | Conner Chen | Meta information modification | 3648 | 2021-04-07 07:39:04 | | |
Video Upload Options
The nuclear factor κB (NF-κB) family are the master transcription factors that control cell proliferation, apoptosis, the expression of interferons and proinflammatory factors, and viral infection. During viral infection, host innate immune system senses viral products, such as viral nucleic acids, to activate innate defense pathways, including the NF-κB signaling axis, thereby inhibiting viral infection. In these NF-κB signaling pathways, diverse types of ubiquitination have been shown to participate in different steps of the signal cascades. Recent advances find that viruses also modulate the ubiquitination in NF-κB signaling pathways to activate viral gene expression or inhibit host NF-κB activation and inflammation, thereby facilitating viral infection. Understanding the role of ubiquitination in NF-κB signaling during viral infection will advance our knowledge of regulatory mechanisms of NF-κB signaling and pave the avenue for potential antiviral therapeutics.
1. NF-κB Signaling Pathways
NF-κB is inactive in resting cells due to the cytosolic sequestration by the inhibitors of κBs (IκBs). The typical IκBs, such as IκBα and IκBβ, block the nuclear localization sequence (NLS) of NF-κB to retain it in the cytoplasm [1]. However, NF-κB is rapidly activated and translocated to the nucleus upon stimulations, such as TNF, viral infection, and UV. The activation of NF-κB is mainly through two distinct signaling branches: the canonical pathway and the noncanonical pathway (Figure 1) [2]. Both pathways are important for controlling viral infection and regulating immune and inflammatory responses. The canonical NF-κB activation is a downstream part of the signaling cascades of many signaling pathways elicited by diverse stimuli, including inflammatory cytokines and pathogen-associated molecular patterns (PAMPs). Although the upper part of these signaling cascades, such as receptors and adaptors, are diverse, they all activate the IκB kinase (IKK) complex consisting of two catalytic subunits, IKKα and IKKβ, and a regulatory subunit named NF-κB essential modulator (NEMO) (also known as IKKγ) [3]. The activated IKK complex further phosphorylates two serines of IκBα at the N-terminus, thereby triggering ubiquitin-dependent IκBα degradation in the proteasome and resulting in the release and translocation of NF-κB into the nucleus. The p50/p65 and p50/c-Rel dimers are the predominant NF-κB transcription factors inducing gene expression in the canonical pathway.
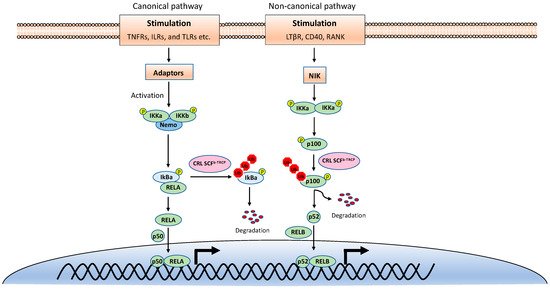
Figure 1. The canonical and noncanonical nuclear factor κB (NF-κB) pathways.
By contrast, the noncanonical NF-κB pathway is only activated by a narrow set of stimuli, such as the ligands of TNFR superfamily members lymphotoxin beta receptor (LT-βR), B-cell activating factor (BAFF), CD40, RANK, and the viral latent membrane protein 1 (LMP1) of Epstein–Barr virus (EBV). Furthermore, unlike the canonical pathway, IκBα degradation is not involved in the noncanonical NF-κB pathway. Instead, the noncanonical NF-κB pathway involves in the processing of the NF-κB2 precursor p100 to the mature form p52 [4]. Upon stimulation, the NF-κB-inducing kinase (NIK) activates IKKα, and then the activated IKKα phosphorylates p100, leading to its polyubiquitination and processing p52 by partial degradation of its C-terminal ankyrin repeats. The p52 forms a dimer with RelB and translocates into the nucleus to induce gene expression. Despite the different mechanisms in canonical and noncanonical NF-κB pathways, they both are regulated by ubiquitination [5][6].
2. Ubiquitination
2.1. Ubiquitination Process
The ubiquitination process is a three-step enzymatic reaction (Figure 2A). First, the E1 enzyme activates ubiquitin by linking the C-terminus of ubiquitin to E1 via a thioester bond in an ATP dependent manner. Second, the activated ubiquitin is transferred to the E2 conjugating enzyme, forming a thioester bond with the activated ubiquitin. Last, the E2 forms a complex with the E3 ligating enzyme, and the E2–E3 enzyme complex conjugates the ubiquitin to the substrate protein through an isopeptide bond between the lysine of the substrate protein and the C-terminal glycine of ubiquitin. Recently, it is found that cysteine, serine and threonine residues, as well as the free amino group of the N-terminus of proteins, also function as sites for ubiquitination through forming thioester, hydroxyester and peptide bonds, respectively [7]. In the human genome, there are two E1s (UBA1 and UBA6), about 35 E2s and more than 600 E3s [8]. The E3 ligases are diverse, and most of them consist of an interacting domain that determines the specificity of the substrate.
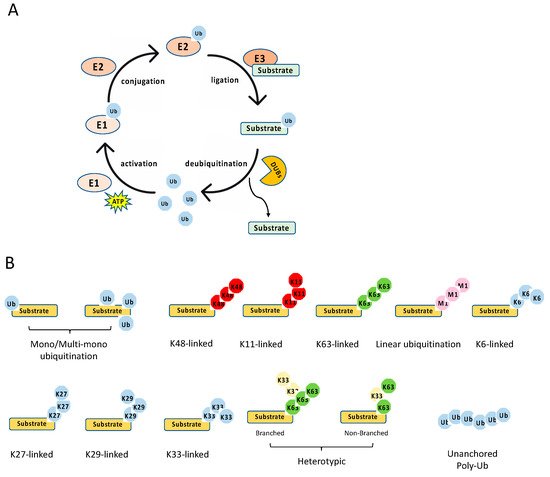
Figure 2. Ubiquitination process and types. (A) The multiple steps of the enzymatic process for ubiquitination controlled by E1, E2, E3, and deubiquitinases (DUBs). (B) The schematics of various types of ubiquitination.
The E3 ligases are subdivided into three distinct classes: the really interesting new gene (RING), the homologous to the E6-associated protein C terminus (HECT), and the RING-between-RING (RBR) [9][10]. The RING E3 ligases are different from HECT and RBR ligases in two aspects. First, RING ligases bind the E2 and the substrate simultaneously and transfer the ubiquitin directly from the E2 to the substrate. By contrast, HECT and RBR ligases form an E3-ubiquitin intermediate via the active site cysteine in these E3 ligases. Then, the ubiquitin is transferred to a substrate of the E3 ligase. Second, in addition to functional monomers, RING ligases form homodimers and heterodimers and can be a part of large multisubunit complexes, such as the cullin-RING ligase (CRL) superfamily.
Ubiquitination is a reversible post-translational modification (Figure 2A). The removal of a ubiquitin or polyubiquitin chain from protein is mediated by deubiquitinases (DUBs), a group of proteases specifically targeting ubiquitin. The human genome has more than fifty DUBs, which are grouped into two main classes: the metalloproteases (JAB1/MPN/Mv34 metalloenzymes (JAMMs)) and the cysteine proteases [ubiquitin-specific protease (USP), ubiquitin C-terminal hydrolases (UCHs), MIU-containing novel DUB family (Mindy), Machado–Joseph disease proteases (MJDs), ovarian tumor proteases (OTUs)] [11]. Like E2 and E3, DUBs also display specificity toward one or a few linkages. For example, the OTU domain-containing deubiquitinase with linear linkage specificity (OTULIN) only hydrolyzes the linear polyubiquitin chain [12][13]. Overall, the E3 and DUBs provide an on–off switch controlling ubiquitination, enabling a regulatory mechanism by external and internal signals.
2.2. Ubiquitination Types
Diverse types of ubiquitination have been found in recent years [9][14] (Figure 2B). First, they can be grouped into mono-ubiquitination and polyubiquitination according to the amount of ubiquitin on the lysine residue of the protein substrate. Mono-ubiquitination is a single ubiquitin molecule conjugated to a lysine of the substrate, whereas polyubiquitination is the conjugation of a ubiquitin polymer to the substrate. Proteins also can be modified at multiple lysine residues with a single ubiquitin molecule, which is called multi-monoubiquitylation. Second, the polyubiquitin chains are grouped into different linkages determined by the conjugation through one of their lysine residues (K6, K11, K27, K29, K33, K48, K63) in the ubiquitin. In addition to these seven chain types, the N-terminal methionine residue (M1) of the ubiquitin can be conjugated to the carboxyl terminus of another ubiquitin forms a Met1-linked polyubiquitin chain (also known as linear polyubiquitination). Third, the polyubiquitin chains can also be classified into homotypic and heterotypic chains. The homotypic chains comprise only a single linkage type. By contrast, heterotypic chains contain mixed linkages within the same polymer. Last, one ubiquitin molecule can be ubiquitinated at two or more sites to form a branched polyubiquitin chain.
The type of ubiquitination determines the fate of the substrate, resulting in different outcomes. For example, the K11- and K48-linked polyubiquitin chains usually drive proteasomal degradation, whereas the monoubiquitylation and K63-linked polyubiquitination mainly involve in protein–protein interaction, DNA damage response, protein kinase activation, and the regulation of protein localization [9]. K27-linked polyubiquitination is involved in the DNA damage response and innate immunity, whereas K29-linked polyubiquitin inhibits Wnt signaling [14]. In addition, K33 linkages have been implicated in post-Golgi protein trafficking [15]. Several types of polyubiquitination tightly regulate NF-κB signaling pathways, including K48- and K63-linked polyubiquitination, Met1-linked polyubiquitination, and unanchored polyubiquitin, which are reviewed in detail below with a focus on the E3 ligases and DUBs involved in NF-κB pathways.
2.2.1. K48-Linked Polyubiquitination
In most scenarios, K48-linked polyubiquitination leads to proteasomal degradation of the target protein. K48-linked polyubiquitination is the first characterized polyubiquitination and plays an essential role in both canonical and noncanonical NF-κB pathways. In the canonical NF-κB pathway, the critical step is the phosphorylation-induced ubiquitination and degradation of IκBs, allowing NF-κB dimers to translocate into the nucleus [2]. Phosphorylation of IκB at serines 32 and 36 creates a degron recognized by the E3 ubiquitin ligase complex comprising Skp, cullin, F-box (SCF) [16][17]. Mechanistically, phosphorylation of IκBs recruits the β-transducing repeat-containing protein (β-TrCP) that bridges an SCF complex consisting of S-phase kinase-associated protein 1 (Skp1), cullin, RING-box protein 1 (Rbx1) and the E2 conjugating enzymes UBCH5b, UBCH5c or UBCH3 to IκB. This complex catalyzes K48-linked polyubiquitination of IκB proteins, resulting in IκB degradation by 26S proteasome and the subsequent release of NF-κB dimers into the nucleus. The K48-linked ubiquitination of IκBs is also reversed by a couple of DUBs. USP11 and USP15 have been shown to deubiquitinate IκB in the TNF-induced NF-κB pathway [18][19]. However, it is not clear whether these DUBs also are involved in other canonical NF-κB pathways.
In the noncanonical NF-κB pathway, the processing of p100 for generating p52 is also dependent on K48-linked polyubiquitination [16][17]. Interestingly, p100 has a similar motif to that one found in IκB proteins. After the motif is phosphorylated by IKKα, p100 is also recognized by the SCF-β-TrCP E3 ligase complex, which leads to partial degradation of the C terminal ankyrin repeats of p100 by the proteasome to make p52. Recent studies further found that p100 can be fully degraded by K48-linked ubiquitination via different sites and E3 ligase. Regarding this complete degradation pathway, the p100 protein is first phosphorylated by glycogen synthase kinase-3 (GSK3). Then, these phosphorylated sites recruit another SCF E3 ligase complex, SCF-Fbw7 [20][21]. The SCF-Fbw7 induces K48-linked polyubiquitination and triggers the complete degradation of p100 by the proteasome.
K48-linked polyubiquitination is the most common type of polyubiquitination and also widely regulates the upstream of all NF-κB pathways, which is discussed in Section 4. In addition, unlike the K63-linked polyubiquitination and linear polyubiquitination discussed below, the E2 conjugating enzymes for K48-linked polyubiquitination are not limited to one or two specific enzymes. Similarly, the E3 ligases for K48-linked polyubiquitination are also diverse and have much less specificity on E2.
2.2.2. K63-Linked Polyubiquitination
In NF-κB pathways, K63-linked polyubiquitin plays a critical role in stabilizing the receptor signalosome on the membrane, facilitating the recruitment of downstream adaptors or complexes, and activating kinases [6]. For example, in the IL-1-induced NF-κB signaling, TRAF6 functions as an E3 ligase to catalyze the synthesis of K63-linked polyubiquitin with the E2 enzyme Ubc13-Uev1A, leading to the activation of the TAK1 kinase complex, which in turn phosphorylates and activates IKK. Many E3 ligases in various NF-κB pathways are capable of catalyzing the K63-linked polyubiquitin; however, it was first thought that Ubc13-Uev1A is the sole E2 enzyme specifically for K63-linked polyubiquitination. Furthermore, genetic studies found that NF-κB activation mediated by TNFα, IL-1β, and TLR ligands is almost normal in several types of cells isolated from Ubc13 knockout mice [22]. Of note, another study showed that IL-1β-induced IKK activation is reduced in Ubc13 knockout mouse embryonic fibroblasts (MEFs) [23]. To reconcile the discrepancy, a study further showed that IL-1-induced NF-κB activation was dependent on Ubc13 while TNFα-induced NF-κB activation was dependent on Ubc5, an E2 enzyme that can synthesize heterogeneous polyubiquitin chains [24].
2.2.3. Met1-Linked Polyubiquitination
Met1-linked or linear polyubiquitination is a new type of ubiquitin modification in NF-kB pathways [25]. Several proteins in NF-κB pathways are the known targets for Met1-linked polyubiquitination, including NEMO, RIP1, FADD, and BCL10. The Met1-linked polyubiquitin chain is assembled by the linear ubiquitin chain assembly complex (LUBAC), a ubiquitin E3 ligase complex comprising HOIL-1 interacting protein (HOIP), heme-oxidized IRP2 ubiquitin ligase 1 (HOIL-1), and SHANK-interacting protein-like 1 (SHARPIN) [26][27][28][29][30] together with the E2 conjugating enzyme UBE2L3 [31]. LUBAC is the only known E3 ligase that catalyzes Met1-linked polyubiquitin; however, LUBAC also mediates K48-linked ubiquitination of TRIM25, an E3 ligase for the cytosolic RNA sensor, retinoic acid-inducible gene I (RIG-I) [32].
2.2.4. Unanchored Polyubiquitin
Studies showed that the unanchored polyubiquitin chains, which are not conjugated to any other cellular proteins, can activate TGFβ-activated kinase 1 (TAK1) in vitro. The unanchored K63-linked polyubiquitin chains directly activate the TAK1 kinase complex in vitro through binding to the TAK1-binding protein 2 (TAB2) or TAB3 subunit of this complex. The activated TAK1 then phosphorylates IKKβ, leading to IKK activation [33]. Similarly, K63-linked polyubiquitin also activates RIG-I in an in vitro reconstitution system [34]. Nonetheless, more work is needed to elucidate protein kinase activation mechanism by unanchored polyubiquitin chains and their roles in vivo.
3. Ubiquitination in NF-κB Signaling Pathways
As mentioned above, many stimuli activate NF-κB pathways and the signal cascades upstream of IKK and NIK are diverse and complex, although they are also regulated by ubiquitination. Thus, this review only focuses on two examples, TNFR1 and RIG-I signaling pathways (Figure 3). TNFR1 is a well-studied inflammatory cytokines-induced signaling pathway, while RIG-I is the major antiviral innate immune pathway in response to RNA virus infection. These pathways are not only critical for inflammation and host defense during viral infection but also are extensively examined for polyubiquitin-mediated regulation.
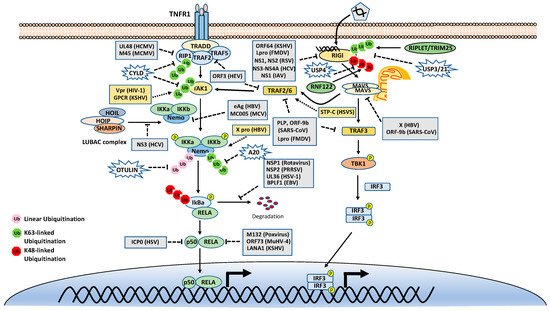
Figure 3. Tumor necrosis factor receptor type 1 (TNFR1) and retinoic acid-inducible gene I (RIG-I) signaling pathways. K48-, K63-, and Met1-linked polyubiquitinations are illustrated. Virus proteins that suppress or activate NF-κB via modulation of ubiquitination are indicated.
3.1. Ubiquitination in the TNFR1 Signaling Pathway
The TNF receptor 1 (TNFR1)-signaling pathway is the most well-studied canonical NF-κB pathway. TNFR1 is a receptor for the inflammatory cytokine, tumor necrosis factor-alpha (TNFα), which induces inflammation, apoptosis, and necrosis. Once engaged with TNFα, TNFR1 trimerizes and recruits different cellular adaptors to form complex I and complex II, leading to NF-κB activation and cell death, respectively. To fit the review topic, we focus on the signaling pathway elicited by TNFR1 complex I. Within the complex I, the tumor necrosis factor receptor type 1-associated death domain protein (TRADD) binds TNFR1 through the death domain. TRADD further recruits the receptor-interacting serine/threonine–protein kinase 1 (RIPK1) and the E3 ligases TNF receptor-associated factor 2 (TRAF2), TRAF5, and c-IAPs. cIAPs or TRAFs generate K63-linked polyubiquitin chains on TRAF2 and RIP1. The K63-linked polyubiquitin chains then recruit the TAK1-TAB complex and the IKK complex to the receptor complexes via K63-selective binding of TAB2/3 or NEMO, respectively. Recent studies also showed that the LUBAC E3 ligase complex is also recruited to the TNFR1 and generates Met1-linked polyubiquitin on RIP1 and NEMO [25]. These Met1-linked polyubiquitin chains also function as a scaffold to recruit the IKK complex via the ubiquitin-binding domain of NEMO. Within the TNFR1 signalosome complex, TAK1 activates the IKK by phosphorylation, and then the activated IKK complex phosphorylates IκBα. The phosphorylated IκBα is ubiquitinated by the E3 complex SCFβ-TrCP for the K48-ubiquitination-mediated proteasomal degradation of IκBα. After liberation from IκBα, the canonical NF-κB transcription factors, predominantly composed of homo- or hetero-dimers of p65 (RelA) and p50, translocate into the nucleus and activate NF-κB target genes (Figure 3).
DUBs are also engaged with a opposite role to E3 ligases by balancing ubiquitination modification. The K63-linked polyubiquitin is hydrolyzed by the cylindromatosis gene CYLD and A20 (also known as TNFα-induced protein 3). CYLD contains a USP domain in the C-terminus, which mediates the cleavage of polyubiquitins. In the NF-κB pathways, CYLD removes the K63-linked polyubiquitin chains from TAK1, RIP1, BCL3, TRAF2, TRAF6, and NEMO. Unlike CYLD, A20 is a hybrid of DUB and E3 ligase, which has an N-terminal OTU domain responsible for polyubiquitin cleavage and a C-terminal domain of seven Cys2-Cys2 zinc-fingers (ZF) that render the E3 ligase activity. A20 disassembles K63-linked polyubiquitin chains of RIP1, TRAF6, RIPK2, NEMO and MALT1, thus inhibiting the activation of NF-κB activation. A20 also mediates K48-linked ubiquitination of RIP1, leading to its degradation, thus inhibits NF-κB activation. The cellular zinc finger anti-NF-κB (Cezanne) is a deubiquitinase of the ovarian tumor superfamily with sequence similarity to A20. Cezanne also suppresses NF-κB activation by targeting RIP1 signaling intermediaries for deubiquitination [35].
In addition, the ubiquitin-specific peptidase 21 (USP21) inhibits NF-κB in the TNF pathway by deubiquitination of RIP1 [36]. USP31 and USP7 also inhibit NF-κB signaling by deubiquitinating K63-linked polyubiquitin chains [37][38]. MCP-induced protein 1 (MCPIP1) (also known as Zc3h12a) deubiquitinates TRAF proteins and negatively regulates JNK and NF-κB signaling [39]. USP11 and USP15 remove K48-linked chains from IκBα, thus protecting IκBα from degradation by the proteasome [18][19]. Interestingly, USP2 positively regulates TNF-induced NF-κB activation; however, the target of USP2 is unknown [40].
The Met1-linked polyubiquitin chain is cleaved by CYLD and OTULIN. CYLD exhibits deubiquitinase activity toward multiple types of polyubiquitin; however, OTULIN specifically targets Met1-linked polyubiquitin. OTULIN also interacts with HOIP via its PUB-interacting motif and inhibits LUBAC activity [41][42]. Recently, we found that TRIM32 conjugates nonproteolytic polyubiquitin onto OTULIN and the polyubiquitin blocks the interaction between HOIP and OTULIN, thereby activating NF-κB signaling [43].
3.2. Ubiquitination in the RIG-I Signaling Pathway
The innate immune system uses pattern recognition receptors (PRRs) in different cellular compartments to sense microbial components that mark invading viruses. Many of these PRRs have been characterized, including the RIG-I-like receptors (RLRs), such as RIG-I, MDA5 and LGP2 [44][45]. The RLRs recognize viral RNA in the cytoplasm, for example, the double-stranded RNA (dsRNA) or 5’ triphosphate RNA generated by viral replication of RNA viruses [46][47][48][49][50][51]. The engagement of viral RNA induces the conformational change of RIG-I and several post-translational modifications, which leads to the oligomerization of RIG-I. The oligomerized RIG-I translocates to the mitochondria and binds the mitochondrial antiviral signaling protein (MAVS, also known as CARDIF, IPS1, and VISA). The binding results in the oligomerization of MAVS and the recruitment of TANK-binding kinase 1 (TBK1). The oligomerized MAVS further recruits TBK1, TRAF6, IKK and interferon regulatory factor 3 (IRF3). Subsequently, TBK1 phosphorylates IRF3, which in turn triggers its dimerization and nuclear translocation [52][53][54][55][56], whereas IKK phosphorylates IκBα to release NF-κB. In the nucleus, IRF3 and NF-κB, together with other transcription factors, form active transcriptional complexes and activate type I IFN expression [52][53][54][55][57] (Figure 3).
Similar to the TNFR1 signaling pathway, the RIG-I signaling pathway is also heavily regulated by ubiquitination. First, RIG-I undergoes several types of ubiquitination, which is regulated by at least nine E3 ligases and four DUBs. Three ubiquitin E3 ligases, TRIM25 [58], MEX3C [59], and TRIM4 [60], have been reported to activate RIG-I signaling by mediating K63-linked polyubiquitination of the N-terminal CARD domain of RIG-I. TRIM25 was first found to bind the CARD and mediate polyubiquitination of CARD at Lys172 [58]. Other studies showed that MEX3C and TRIM4 target RIG-I for ubiquitination at different sites [59][60]. Recently, a study showed that another ubiquitin E3 ligase, RIPLET (a.k.a. REUL), is the predominant E3 ligase for RIG-I K63-linked ubiquitination and activation [61][62][63]; however, RIPLET ubiquitinates the CTD domain of RIG-I at Lys849 and Lys851 [64]. Interestingly, unanchored K63-linked polyubiquitin is also found to non-covalently bind the CARD and promote CARD tetramerization and concomitant signal activation [65]. These activating K63-linked polyubiquitin chains of RIG-I can be removed by several DUBs, including USP3, USP21 and CYLD [66][67][68][69]. RIG-I also can be conjugated with K48-linked polyubiquitin. The E3 ligases RNF122, RNF125, c-Cbl, and CHIP have been found to mediate K48-linked ubiquitination and proteasomal degradation of RIG-I, thereby inhibiting RIG-I signaling [70][71][72][73]. Another E3 ligase TRIM40 conjugates both K27- and K48-linked polyubiquitin onto RIG-I for proteasomal degradation [74]. The K48-linked ubiquitination of RIG-I is reversed by USP4 [75]. In addition, LUBAC mediates K48-linked ubiquitination of TRIM25, an E3 ligase for RIG-I K63-linked ubiquitination and activation. The K48-linked ubiquitination leads to TRIM25 protein degradation, thereby inhibiting RIG-I activation [32]. In contrast, USP15 deubiquitylates TRIM25, preventing the LUBAC-dependent degradation of TRIM25 [76].
The downstream of RIG-I is also regulated by ubiquitination. As with RIG-I, MAVS is also ubiquitinated multiple E3 ligases, although most of them mediate K48-linked ubiquitination and proteasomal degradation. Two HEC domain-containing E3 ligases, the Smad ubiquitin regulatory factor 1 (Smurf1) and Smurf2, also mediate K48-linked ubiquitination of MAVS [77][78]. Other E3 ligases, including MARCH5, RNF5, AIP4, TRIM25, and pVHL, are all reported to promote MAVS protein degradation through K48-linked polyubiquitination [79][80][81][82]. Unlike the redundancy of E3 ligases for K48-linked polyubiquitination, there is only one E3 ligase TRIM31 was reported to confer the K63-linked polyubiquitin onto MAVS and promotes MAVS aggregation and activation [83]. TRIM21 promotes innate immune response to RNA viral infection through K27-linked polyubiquitination of MAVS [84]. There are two DUBs for MAVS, the ovarian tumor family deubiquitinase 4 (OTUD4) and YOD1, cleaves K48- and K63-linked polyubiquitin chains of MAVS, respectively [85][86]. Interestingly, TRIM44, an unusual DUB, stabilizes MAVS by preventing its ubiquitination and degradation [87]. In addition, K63-linked ubiquitination of TRIM14 bridges NEMO to MAVS [88].
Interestingly, two E3 ligases catalyze non-K48-linked polyubiquitin chains on MAVS to promote its autophagic degradation. The E3 ubiquitin ligase MARCH8 catalyzes the K27-linked polyubiquitin chain on MAVS at lysine 7, which serves as a recognition signal for NDP52-dependent autophagic degradation [89]. Another E3 ligase RNF34 initiates the K63- to K27-linked polyubiquitin switch on MAVS at Lys 311, thus facilitating the autophagic degradation of MAVS [90].
TBK1 is the key kinase for the IRF3 signaling axis of the RIG-I pathway. We and others showed that TBK1 is K63-linked ubiquitinated for activation by E3 ligases mind bomb 1 (MIB1), MIB2, and RNF128 [91][92]. Overall, the RIG-I signaling pathway is heavily regulated by different types of ubiquitination, which provides multiple layers of regulation for this pathway.
References
- Henkel, T.; Zabel, U.; van Zee, K.; Muller, J.M.; Fanning, E.; Baeuerle, P.A. Intramolecular masking of the nuclear location signal and dimerization domain in the precursor for the p50 NF-kappa B subunit. Cell 1992, 68, 1121–1133.
- Hayden, M.S.; Ghosh, S. NF-kappaB, the first quarter-century: Remarkable progress and outstanding questions. Genes Dev. 2012, 26, 203–234.
- Hinz, M.; Scheidereit, C. The IkappaB kinase complex in NF-kappaB regulation and beyond. EMBO Rep. 2014, 15, 46–61.
- Sun, S.C. The non-canonical NF-kappaB pathway in immunity and inflammation. Nat. Rev. Immunol. 2017, 17, 545–558.
- Iwai, K. Diverse ubiquitin signaling in NF-kappaB activation. Trends Cell Biol. 2012, 22, 355–364.
- Chen, J.; Chen, Z.J. Regulation of NF-kappaB by ubiquitination. Curr. Opin. Immunol. 2013, 25, 4–12.
- McClellan, A.J.; Laugesen, S.H.; Ellgaard, L. Cellular functions and molecular mechanisms of non-lysine ubiquitination. Open Biol. 2019, 9, 190147.
- Berndsen, C.E.; Wolberger, C. New insights into ubiquitin E3 ligase mechanism. Nat. Struct. Mol. Biol. 2014, 21, 301–307.
- Komander, D.; Rape, M. The Ubiquitin Code. Annu. Rev. Biochem. 2012, 81, 203–229.
- Collins, P.E.; Mitxitorena, I.; Carmody, R.J. The Ubiquitination of NF-kappaB Subunits in the Control of Transcription. Cells 2016, 5, 23.
- Nijman, S.M.; Luna-Vargas, M.P.; Velds, A.; Brummelkamp, T.R.; Dirac, A.M.; Sixma, T.K.; Bernards, R. A genomic and functional inventory of deubiquitinating enzymes. Cell 2005, 123, 773–786.
- Fiil, B.K.; Damgaard, R.B.; Wagner, S.A.; Keusekotten, K.; Fritsch, M.; Bekker-Jensen, S.; Mailand, N.; Choudhary, C.; Komander, D.; Gyrd-Hansen, M. OTULIN restricts Met1-linked ubiquitination to control innate immune signaling. Mol. Cell 2013, 50, 818–830.
- Keusekotten, K.; Elliott, P.R.; Glockner, L.; Fiil, B.K.; Damgaard, R.B.; Kulathu, Y.; Wauer, T.; Hospenthal, M.K.; Gyrd-Hansen, M.; Krappmann, D.; et al. OTULIN antagonizes LUBAC signaling by specifically hydrolyzing Met1-linked polyubiquitin. Cell 2013, 153, 1312–1326.
- Akutsu, M.; Dikic, I.; Bremm, A. Ubiquitin chain diversity at a glance. J. Cell Sci. 2016, 129, 875–880.
- Yuan, W.C.; Lee, Y.R.; Lin, S.Y.; Chang, L.Y.; Tan, Y.P.; Hung, C.C.; Kuo, J.C.; Liu, C.H.; Lin, M.Y.; Xu, M.; et al. K33-Linked Polyubiquitination of Coronin 7 by Cul3-KLHL20 Ubiquitin E3 Ligase Regulates Protein Trafficking. Mol. Cell 2014, 54, 586–600.
- Spencer, E.; Jiang, J.; Chen, Z.J. Signal-induced ubiquitination of IkappaBalpha by the F-box protein Slimb/beta-TrCP. Genes Dev. 1999, 13, 284–294.
- Winston, J.T.; Strack, P.; Beer-Romero, P.; Chu, C.Y.; Elledge, S.J.; Harper, J.W. The SCFbeta-TRCP-ubiquitin ligase complex associates specifically with phosphorylated destruction motifs in IkappaBalpha and beta-catenin and stimulates IkappaBalpha ubiquitination in vitro. Genes Dev. 1999, 13, 270–283.
- Schweitzer, K.; Bozko, P.M.; Dubiel, W.; Naumann, M. CSN controls NF-kappaB by deubiquitinylation of IkappaBalpha. EMBO J. 2007, 26, 1532–1541.
- Sun, W.; Tan, X.; Shi, Y.; Xu, G.; Mao, R.; Gu, X.; Fan, Y.; Yu, Y.; Burlingame, S.; Zhang, H.; et al. USP11 negatively regulates TNFalpha-induced NF-kappaB activation by targeting on IkappaBalpha. Cell Signal. 2010, 22, 386–394.
- Fukushima, H.; Matsumoto, A.; Inuzuka, H.; Zhai, B.; Lau, A.W.; Wan, L.; Gao, D.; Shaik, S.; Yuan, M.; Gygi, S.P.; et al. SCF(Fbw7) modulates the NFkB signaling pathway by targeting NFkB2 for ubiquitination and destruction. Cell Rep. 2012, 1, 434–443.
- Busino, L.; Millman, S.E.; Scotto, L.; Kyratsous, C.A.; Basrur, V.; O’Connor, O.; Hoffmann, A.; Elenitoba-Johnson, K.S.; Pagano, M. Fbxw7alpha- and GSK3-mediated degradation of p100 is a pro-survival mechanism in multiple myeloma. Nat. Cell Biol. 2012, 14, 375–385.
- Yamamoto, M.; Okamoto, T.; Takeda, K.; Sato, S.; Sanjo, H.; Uematsu, S.; Saitoh, T.; Yamamoto, N.; Sakurai, H.; Ishii, K.J.; et al. Key function for the Ubc13 E2 ubiquitin-conjugating enzyme in immune receptor signaling. Nat. Immunol. 2006, 7, 962–970.
- Yamazaki, K.; Gohda, J.; Kanayama, A.; Miyamoto, Y.; Sakurai, H.; Yamamoto, M.; Akira, S.; Hayashi, H.; Su, B.; Inoue, J. Two mechanistically and temporally distinct NF-kappaB activation pathways in IL-1 signaling. Sci. Signal. 2009, 2, ra66.
- Xu, M.; Skaug, B.; Zeng, W.; Chen, Z.J. A ubiquitin replacement strategy in human cells reveals distinct mechanisms of IKK activation by TNFalpha and IL-1beta. Mol. Cell 2009, 36, 302–314.
- Iwai, K.; Tokunaga, F. Linear polyubiquitination: A new regulator of NF-kappaB activation. EMBO Rep. 2009, 10, 706–713.
- Haas, T.L.; Emmerich, C.H.; Gerlach, B.; Schmukle, A.C.; Cordier, S.M.; Rieser, E.; Feltham, R.; Vince, J.; Warnken, U.; Wenger, T.; et al. Recruitment of the linear ubiquitin chain assembly complex stabilizes the TNF-R1 signaling complex and is required for TNF-mediated gene induction. Mol. Cell 2009, 36, 831–844.
- Tokunaga, F.; Sakata, S.; Saeki, Y.; Satomi, Y.; Kirisako, T.; Kamei, K.; Nakagawa, T.; Kato, M.; Murata, S.; Yamaoka, S.; et al. Involvement of linear polyubiquitylation of NEMO in NF-kappaB activation. Nat. Cell Biol. 2009, 11, 123–132.
- Tokunaga, F.; Nakagawa, T.; Nakahara, M.; Saeki, Y.; Taniguchi, M.; Sakata, S.; Tanaka, K.; Nakano, H.; Iwai, K. SHARPIN is a component of the NF-kappaB-activating linear ubiquitin chain assembly complex. Nature 2011, 471, 633–636.
- Ikeda, F.; Deribe, Y.L.; Skanland, S.S.; Stieglitz, B.; Grabbe, C.; Franz-Wachtel, M.; van Wijk, S.J.; Goswami, P.; Nagy, V.; Terzic, J.; et al. SHARPIN forms a linear ubiquitin ligase complex regulating NF-kappaB activity and apoptosis. Nature 2011, 471, 637–641.
- Gerlach, B.; Cordier, S.M.; Schmukle, A.C.; Emmerich, C.H.; Rieser, E.; Haas, T.L.; Webb, A.I.; Rickard, J.A.; Anderton, H.; Wong, W.W.; et al. Linear ubiquitination prevents inflammation and regulates immune signalling. Nature 2011, 471, 591–596.
- Fu, B.; Li, S.; Wang, L.; Berman, M.A.; Dorf, M.E. The ubiquitin conjugating enzyme UBE2L3 regulates TNFalpha-induced linear ubiquitination. Cell Res. 2014, 24, 376–379.
- Inn, K.S.; Gack, M.U.; Tokunaga, F.; Shi, M.; Wong, L.Y.; Iwai, K.; Jung, J.U. Linear ubiquitin assembly complex negatively regulates RIG-I- and TRIM25-mediated type I interferon induction. Mol. Cell 2011, 41, 354–365.
- Xia, Z.P.; Sun, L.; Chen, X.; Pineda, G.; Jiang, X.; Adhikari, A.; Zeng, W.; Chen, Z.J. Direct activation of protein kinases by unanchored polyubiquitin chains. Nature 2009, 461, 114–119.
- Zeng, W.; Sun, L.; Jiang, X.; Chen, X.; Hou, F.; Adhikari, A.; Xu, M.; Chen, Z.J. Reconstitution of the RIG-I pathway reveals a signaling role of unanchored polyubiquitin chains in innate immunity. Cell 2010, 141, 315–330.
- Enesa, K.; Zakkar, M.; Chaudhury, H.; Luong, L.A.; Rawlinson, L.; Mason, J.C.; Haskard, D.O.; Dean, J.L.; Evans, P.C. NF-kappaB suppression by the deubiquitinating enzyme Cezanne: A novel negative feedback loop in pro-inflammatory signaling. J. Biol. Chem. 2008, 283, 7036–7045.
- Xu, G.; Tan, X.; Wang, H.; Sun, W.; Shi, Y.; Burlingame, S.; Gu, X.; Cao, G.; Zhang, T.; Qin, J.; et al. Ubiquitin-specific peptidase 21 inhibits tumor necrosis factor alpha-induced nuclear factor kappaB activation via binding to and deubiquitinating receptor-interacting protein 1. J. Biol. Chem. 2010, 285, 969–978.
- Tzimas, C.; Michailidou, G.; Arsenakis, M.; Kieff, E.; Mosialos, G.; Hatzivassiliou, E.G. Human ubiquitin specific protease 31 is a deubiquitinating enzyme implicated in activation of nuclear factor-kappaB. Cell Signal. 2006, 18, 83–92.
- Daubeuf, S.; Singh, D.; Tan, Y.; Liu, H.; Federoff, H.J.; Bowers, W.J.; Tolba, K. HSV ICP0 recruits USP7 to modulate TLR-mediated innate response. Blood 2009, 113, 3264–3275.
- Liang, J.; Saad, Y.; Lei, T.; Wang, J.; Qi, D.; Yang, Q.; Kolattukudy, P.E.; Fu, M. MCP-induced protein 1 deubiquitinates TRAF proteins and negatively regulates JNK and NF-kappaB signaling. J. Exp. Med. 2010, 207, 2959–2973.
- Metzig, M.; Nickles, D.; Falschlehner, C.; Lehmann-Koch, J.; Straub, B.K.; Roth, W.; Boutros, M. An RNAi screen identifies USP2 as a factor required for TNF-alpha-induced NF-kappaB signaling. Int. J. Cancer 2011, 129, 607–618.
- Elliott, P.R.; Nielsen, S.V.; Marco-Casanova, P.; Fiil, B.K.; Keusekotten, K.; Mailand, N.; Freund, S.M.; Gyrd-Hansen, M.; Komander, D. Molecular basis and regulation of OTULIN-LUBAC interaction. Mol. Cell 2014, 54, 335–348.
- Schaeffer, V.; Akutsu, M.; Olma, M.H.; Gomes, L.C.; Kawasaki, M.; Dikic, I. Binding of OTULIN to the PUB domain of HOIP controls NF-kappaB signaling. Mol. Cell 2014, 54, 349–361.
- Zhao, M.; Song, K.; Hao, W.; Wang, L.; Patil, G.; Li, Q.; Xu, L.; Hua, F.; Fu, B.; Schwamborn, J.C.; et al. Non-proteolytic ubiquitination of OTULIN regulates NF-kappaB signaling pathway. J. Mol. Cell Biol. 2019.
- Takeuchi, O.; Akira, S. Pattern recognition receptors and inflammation. Cell 2010, 140, 805–820.
- Wilkins, C.; Gale, M., Jr. Recognition of viruses by cytoplasmic sensors. Curr. Opin. Immunol. 2010, 22, 41–47.
- Pichlmair, A.; Schulz, O.; Tan, C.P.; Naslund, T.I.; Liljestrom, P.; Weber, F.; Reis e Sousa, C. RIG-I-mediated antiviral responses to single-stranded RNA bearing 5′-phosphates. Science 2006, 314, 997–1001.
- Hornung, V.; Ellegast, J.; Kim, S.; Brzozka, K.; Jung, A.; Kato, H.; Poeck, H.; Akira, S.; Conzelmann, K.K.; Schlee, M.; et al. 5’-Triphosphate RNA is the ligand for RIG-I. Science 2006, 314, 994–997.
- Yoneyama, M.; Kikuchi, M.; Natsukawa, T.; Shinobu, N.; Imaizumi, T.; Miyagishi, M.; Taira, K.; Akira, S.; Fujita, T. The RNA helicase RIG-I has an essential function in double-stranded RNA-induced innate antiviral responses. Nat. Immunol. 2004, 5, 730–737.
- Kang, D.C.; Gopalkrishnan, R.V.; Wu, Q.; Jankowsky, E.; Pyle, A.M.; Fisher, P.B. mda-5: An interferon-inducible putative RNA helicase with double-stranded RNA-dependent ATPase activity and melanoma growth-suppressive properties. Proc. Natl. Acad. Sci. USA 2002, 99, 637–642.
- Kato, H.; Takeuchi, O.; Sato, S.; Yoneyama, M.; Yamamoto, M.; Matsui, K.; Uematsu, S.; Jung, A.; Kawai, T.; Ishii, K.J.; et al. Differential roles of MDA5 and RIG-I helicases in the recognition of RNA viruses. Nature 2006, 441, 101–105.
- Cui, S.; Eisenacher, K.; Kirchhofer, A.; Brzozka, K.; Lammens, A.; Lammens, K.; Fujita, T.; Conzelmann, K.K.; Krug, A.; Hopfner, K.P. The C-terminal regulatory domain is the RNA 5’-triphosphate sensor of RIG-I. Mol. Cell 2008, 29, 169–179.
- Sharma, S.; tenOever, B.R.; Grandvaux, N.; Zhou, G.P.; Lin, R.; Hiscott, J. Triggering the interferon antiviral response through an IKK-related pathway. Science 2003, 300, 1148–1151.
- Fitzgerald, K.A.; McWhirter, S.M.; Faia, K.L.; Rowe, D.C.; Latz, E.; Golenbock, D.T.; Coyle, A.J.; Liao, S.M.; Maniatis, T. IKKepsilon and TBK1 are essential components of the IRF3 signaling pathway. Nat. Immunol. 2003, 4, 491–496.
- Hemmi, H.; Takeuchi, O.; Sato, S.; Yamamoto, M.; Kaisho, T.; Sanjo, H.; Kawai, T.; Hoshino, K.; Takeda, K.; Akira, S. The roles of two IkappaB kinase-related kinases in lipopolysaccharide and double stranded RNA signaling and viral infection. J. Exp. Med. 2004, 199, 1641–1650.
- McWhirter, S.M.; Fitzgerald, K.A.; Rosains, J.; Rowe, D.C.; Golenbock, D.T.; Maniatis, T. IFN-regulatory factor 3-dependent gene expression is defective in Tbk1-deficient mouse embryonic fibroblasts. Proc. Natl. Acad. Sci. USA 2004, 101, 233–238.
- Tanaka, Y.; Chen, Z.J. STING specifies IRF3 phosphorylation by TBK1 in the cytosolic DNA signaling pathway. Sci. Signal. 2012, 5, ra20.
- Takeuchi, O.; Akira, S. Innate immunity to virus infection. Immunol. Rev. 2009, 227, 75–86.
- Gack, M.U.; Shin, Y.C.; Joo, C.H.; Urano, T.; Liang, C.; Sun, L.; Takeuchi, O.; Akira, S.; Chen, Z.; Inoue, S.; et al. TRIM25 RING-finger E3 ubiquitin ligase is essential for RIG-I-mediated antiviral activity. Nature 2007, 446, 916–920.
- Kuniyoshi, K.; Takeuchi, O.; Pandey, S.; Satoh, T.; Iwasaki, H.; Akira, S.; Kawai, T. Pivotal role of RNA-binding E3 ubiquitin ligase MEX3C in RIG-I-mediated antiviral innate immunity. Proc. Natl. Acad. Sci. USA 2014, 111, 5646–5651.
- Yan, J.; Li, Q.; Mao, A.P.; Hu, M.M.; Shu, H.B. TRIM4 modulates type I interferon induction and cellular antiviral response by targeting RIG-I for K63-linked ubiquitination. J. Mol. Cell Biol. 2014, 6, 154–163.
- Cadena, C.; Ahmad, S.; Xavier, A.; Willemsen, J.; Park, S.; Park, J.W.; Oh, S.W.; Fujita, T.; Hou, F.; Binder, M.; et al. Ubiquitin-Dependent and -Independent Roles of E3 Ligase RIPLET in Innate Immunity. Cell 2019, 177, 1187–1200.e1116.
- Oshiumi, H.; Miyashita, M.; Inoue, N.; Okabe, M.; Matsumoto, M.; Seya, T. The Ubiquitin Ligase Riplet Is Essential for RIG-I-Dependent Innate Immune Responses to RNA Virus Infection. Cell Host Microbe 2010, 8, 496–509.
- Hayman, T.J.; Hsu, A.C.; Kolesnik, T.B.; Dagley, L.F.; Willemsen, J.; Tate, M.D.; Baker, P.J.; Kershaw, N.J.; Kedzierski, L.; Webb, A.I.; et al. RIPLET, and not TRIM25, is required for endogenous RIG-I-dependent antiviral responses. Immunol. Cell Biol. 2019, 97, 840–852.
- Oshiumi, H.; Matsumoto, M.; Hatakeyama, S.; Seya, T. Riplet/RNF135, a RING finger protein, ubiquitinates RIG-I to promote interferon-beta induction during the early phase of viral infection. J. Biol Chem 2009, 284, 807–817.
- Jiang, X.; Kinch, L.N.; Brautigam, C.A.; Chen, X.; Du, F.; Grishin, N.V.; Chen, Z.J. Ubiquitin-induced oligomerization of the RNA sensors RIG-I and MDA5 activates antiviral innate immune response. Immunity 2012, 36, 959–973.
- Cui, J.; Song, Y.; Li, Y.; Zhu, Q.; Tan, P.; Qin, Y.; Wang, H.Y.; Wang, R.F. USP3 inhibits type I interferon signaling by deubiquitinating RIG-I-like receptors. Cell Res. 2014, 24, 400–416.
- Fan, Y.; Mao, R.; Yu, Y.; Liu, S.; Shi, Z.; Cheng, J.; Zhang, H.; An, L.; Zhao, Y.; Xu, X.; et al. USP21 negatively regulates antiviral response by acting as a RIG-I deubiquitinase. J. Exp. Med. 2014, 211, 313–328.
- Friedman, C.S.; O’Donnell, M.A.; Legarda-Addison, D.; Ng, A.; Cardenas, W.B.; Yount, J.S.; Moran, T.M.; Basler, C.F.; Komuro, A.; Horvath, C.M.; et al. The tumour suppressor CYLD is a negative regulator of RIG-I-mediated antiviral response. Embo Rep. 2008, 9, 930–936.
- Lin, W.; Zhang, J.; Lin, H.Y.; Li, Z.X.; Sun, X.F.; Xin, D.; Yang, M.; Sun, L.W.; Li, L.; Wang, H.M.; et al. Syndecan-4 negatively regulates antiviral signalling by mediating RIG-I deubiquitination via CYLD. Nat. Commun. 2016, 7.
- Wang, W.D.; Jiang, M.H.; Liu, S.; Zhang, S.K.; Liu, W.; Ma, Y.W.; Zhang, L.F.; Zhang, J.Y.; Cao, X.T. RNF122 suppresses antiviral type I interferon production by targeting RIG-I CARDs to mediate RIG-I degradation. Proc. Natl. Acad. Sci. USA 2016, 113, 9581–9586.
- Arimoto, K.I.; Takahashi, H.; Hishiki, T.; Konishi, H.; Fujita, T.; Shimotohno, K. Negative regulation of the RIG-I signaling by the ubiquitin ligase RNF125. Proc. Natl. Acad. Sci. USA 2007, 104, 7500–7505.
- Zhao, K.; Zhang, Q.; Li, X.; Zhao, D.; Liu, Y.; Shen, Q.; Yang, M.; Wang, C.; Li, N.; Cao, X. Cytoplasmic STAT4 Promotes Antiviral Type I IFN Production by Blocking CHIP-Mediated Degradation of RIG-I. J. Immunol. 2016, 196, 1209–1217.
- Chen, W.; Han, C.; Xie, B.; Hu, X.; Yu, Q.; Shi, L.; Wang, Q.; Li, D.; Wang, J.; Zheng, P.; et al. Induction of Siglec-G by RNA viruses inhibits the innate immune response by promoting RIG-I degradation. Cell 2013, 152, 467–478.
- Zhao, C.; Jia, M.; Song, H.; Yu, Z.; Wang, W.; Li, Q.; Zhang, L.; Zhao, W.; Cao, X. The E3 Ubiquitin Ligase TRIM40 Attenuates Antiviral Immune Responses by Targeting MDA5 and RIG-I. Cell Rep. 2017, 21, 1613–1623.
- Wang, L.J.; Zhao, W.; Zhang, M.; Wang, P.; Zhao, K.; Zhao, X.Y.; Yang, S.R.; Gao, C.J. USP4 Positively Regulates RIG-I-Mediated Antiviral Response through Deubiquitination and Stabilization of RIG-I. J. Virol. 2013, 87, 4507–4515.
- Pauli, E.K.; Chan, Y.K.; Davis, M.E.; Gableske, S.; Wang, M.K.; Feister, K.F.; Gack, M.U. The ubiquitin-specific protease USP15 promotes RIG-I-mediated antiviral signaling by deubiquitylating TRIM25. Sci. Signal. 2014, 7, ra3.
- Wang, Y.; Tong, X.; Ye, X. Ndfip1 negatively regulates RIG-I-dependent immune signaling by enhancing E3 ligase Smurf1-mediated MAVS degradation. J. Immunol. 2012, 189, 5304–5313.
- Pan, Y.; Li, R.; Meng, J.L.; Mao, H.T.; Zhang, Y.; Zhang, J. Smurf2 negatively modulates RIG-I-dependent antiviral response by targeting VISA/MAVS for ubiquitination and degradation. J. Immunol. 2014, 192, 4758–4764.
- Yoo, Y.S.; Park, Y.Y.; Kim, J.H.; Cho, H.; Kim, S.H.; Lee, H.S.; Kim, T.H.; Sun Kim, Y.; Lee, Y.; Kim, C.J.; et al. The mitochondrial ubiquitin ligase MARCH5 resolves MAVS aggregates during antiviral signalling. Nat. Commun. 2015, 6, 7910.
- Castanier, C.; Zemirli, N.; Portier, A.; Garcin, D.; Bidere, N.; Vazquez, A.; Arnoult, D. MAVS ubiquitination by the E3 ligase TRIM25 and degradation by the proteasome is involved in type I interferon production after activation of the antiviral RIG-I-like receptors. BMC Biol. 2012, 10, 44.
- You, F.; Sun, H.; Zhou, X.; Sun, W.; Liang, S.; Zhai, Z.; Jiang, Z. PCBP2 mediates degradation of the adaptor MAVS via the HECT ubiquitin ligase AIP4. Nat. Immunol. 2009, 10, 1300–1308.
- Du, J.; Zhang, D.; Zhang, W.; Ouyang, G.; Wang, J.; Liu, X.; Li, S.; Ji, W.; Liu, W.; Xiao, W. pVHL Negatively Regulates Antiviral Signaling by Targeting MAVS for Proteasomal Degradation. J. Immunol. 2015, 195, 1782–1790.
- Liu, B.; Zhang, M.; Chu, H.; Zhang, H.; Wu, H.; Song, G.; Wang, P.; Zhao, K.; Hou, J.; Wang, X.; et al. The ubiquitin E3 ligase TRIM31 promotes aggregation and activation of the signaling adaptor MAVS through Lys63-linked polyubiquitination. Nat. Immunol. 2017, 18, 214–224.
- Xue, B.; Li, H.; Guo, M.; Wang, J.; Xu, Y.; Zou, X.; Deng, R.; Li, G.; Zhu, H. TRIM21 Promotes Innate Immune Response to RNA Viral Infection through Lys27-Linked Polyubiquitination of MAVS. J. Virol. 2018, 92.
- Liuyu, T.; Yu, K.; Ye, L.; Zhang, Z.; Zhang, M.; Ren, Y.; Cai, Z.; Zhu, Q.; Lin, D.; Zhong, B. Induction of OTUD4 by viral infection promotes antiviral responses through deubiquitinating and stabilizing MAVS. Cell Res. 2019, 29, 67–79.
- Liu, C.; Huang, S.; Wang, X.; Wen, M.; Zheng, J.; Wang, W.; Fu, Y.; Tian, S.; Li, L.; Li, Z.; et al. The Otubain YOD1 Suppresses Aggregation and Activation of the Signaling Adaptor MAVS through Lys63-Linked Deubiquitination. J. Immunol. 2019, 202, 2957–2970.
- Yang, B.; Wang, J.; Wang, Y.; Zhou, H.; Wu, X.; Tian, Z.; Sun, B. Novel function of Trim44 promotes an antiviral response by stabilizing VISA. J. Immunol. 2013, 190, 3613–3619.
- Zhou, Z.; Jia, X.; Xue, Q.; Dou, Z.; Ma, Y.; Zhao, Z.; Jiang, Z.; He, B.; Jin, Q.; Wang, J. TRIM14 is a mitochondrial adaptor that facilitates retinoic acid-inducible gene-I-like receptor-mediated innate immune response. Proc. Natl. Acad. Sci. USA 2014, 111, E245.
- Jin, S.; Tian, S.; Luo, M.; Xie, W.; Liu, T.; Duan, T.; Wu, Y.; Cui, J. Tetherin Suppresses Type I Interferon Signaling by Targeting MAVS for NDP52-Mediated Selective Autophagic Degradation in Human Cells. Mol. Cell 2017, 68, 308–322 e304.
- He, X.; Zhu, Y.; Zhang, Y.; Geng, Y.; Gong, J.; Geng, J.; Zhang, P.; Zhang, X.; Liu, N.; Peng, Y.; et al. RNF34 functions in immunity and selective mitophagy by targeting MAVS for autophagic degradation. EMBO J. 2019, 38, e100978.
- Li, S.; Wang, L.; Berman, M.; Kong, Y.Y.; Dorf, M.E. Mapping a dynamic innate immunity protein interaction network regulating type I interferon production. Immunity 2011, 35, 426–440.
- Song, G.; Liu, B.; Li, Z.; Wu, H.; Wang, P.; Zhao, K.; Jiang, G.; Zhang, L.; Gao, C. E3 ubiquitin ligase RNF128 promotes innate antiviral immunity through K63-linked ubiquitination of TBK1. Nat. Immunol. 2016, 17, 1342–1351.



