 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Günter Emons | + 1425 word(s) | 1425 | 2021-03-24 07:48:28 | | | |
| 2 | Catherine Yang | Meta information modification | 1425 | 2021-04-06 12:56:00 | | |
Video Upload Options
GnRH receptors are expressed in other reproductive organs, such as the ovary and in tumors originating from the ovary. In ovarian cancer, GnRH is involved in the regulation of proliferation and metastasis. The effects on ovarian tumors can be indirect or direct. GnRH acts indirectly via the HPG axis and directly via GnRH receptors on the surface of ovarian cancer cells.
1. Introduction
Ovarian cancer is a hormone-dependent disease that is influenced by hormonal signaling pathways [1][2]. This effect asserts itself after the illness and could therefore also have therapeutic potential. Gonadotropin-releasing hormone (GnRH) is pulsatile released from neurons in the hypothalamus and induces expression and pulsatile release of follicle-stimulating hormone (FSH) and luteinizing hormone (LH) from anterior pituitary. Both, FSH and LH in turn promote maturation of follicles, ovulation, corpus luteum formation and synthesis of estrogen and progesterone. In the following, we give an overview on the role of GnRH, the key player in the regulation of ovarian function, in ovarian cancer development and progression.
2. Direct Effects of GnRH on Ovarian Cancer
Time- and dose-dependent anti-proliferative actions of GnRH agonists have been observed in many cell lines from different cancer entities of the reproductive tract. This includes ovarian cancer [3][4][5][6][7]. GnRH antagonists also show significant growth-inhibiting actions on most GnRH receptor-positive cancer cell lines [3][4]. This indicates that GnRH agonists and antagonists may not be differentiated in the GnRH system in cancer cells. In addition, the signaling mechanisms of the GnRH receptor known to be activated in gonadotropic cells of the pituitary, are not connected in the transmission of the proliferation inhibiting actions of GnRH analogs in cancer cells. In pituitary gonadotrophs, specific binding of GnRH to its receptor leads to a conformational change and subsequent activation of the heterotrimeric G-protein subunit αq/11, which leads to calcium-dependent signal transduction [8][9]. Activation of phospholipase C (PLC) causes the hydrolysis of phosphatidylinositol-4,5-bisphosphate (PIP2). This creates diacylglycerol (DAG) and inositol-1,4,5-trisphosphate (IP3) [10][11][12]. IP3 diffuses from the plasma membrane and activates receptors on the endoplasmic reticulum (ER). This leads to an outflow of calcium from the ER into the cytosol [13][14]. DAG remains in the membrane and induces Ca2+-dependent protein kinase C (PKC), which in the course of the induction of mitogen-activated protein kinases (MAPK) [15][16] leads to gondotropin synthesis (LH and FSH) and secretion [15][17][18][19]. The signaling found to be activated by GnRH agonists in the pituitary is not turned on in cancers of the ovary, although PLC, PKC and AC can be activated in these cells by pharmacologic stimulation. [5][20].
In contrast to the pituitary gland, the GnRH receptor-mediated signal transduction in gynecological tumors takes place via coupling with G-protein αi instead of G-protein αq, which may result in variable receptor conformations and signaling complexes [20][21][22] (Figure 1). Since the gene sequence of GnRH receptors in human carcinomas of the ovary is the same as that in the pituitary gland, different receptor conformations may explain why GnRH receptors in ovarian cancer show different mechanisms compared with cells derived from the pituitary. After binding to G-protein αi, the activated GnRH receptor induces a phosphotyrosine phosphatase (PTP) and prevents the signaling of growth factor receptors, which leads to a reduction in proliferation of cancer cells [5][20][23][24][25][26]. Growth factor-driven mitogenic signal transduction is prevented which leads to downregulation of growth factor-mediated activation of mitogen-activated protein kinase (MAPK) [5], c-fos expression [27], and growth factor-induced proliferation [28]. These findings are in agreement with other research on GnRH analogs showing reduction of growth factor receptor expression [29][30][31] and/or growth factor-induced tyrosine kinase activity [5][23][24][26][30][32][33][34]. Induction of apoptosis does not appear to be involved in the down-regulation of cancer cell proliferation by GnRH agonists [3]. In contrast, Imai et al. have reported, that GnRH agonist leuprorelin stimulated intratumoral expression of apoptosis-inducing Fas ligand in GnRH receptor-positive tumor cells. At a concentration of 10 µM, leuprorelin induced up to a 90% reduction in cell number preceded by Fas ligand production [35][36][37][38]. However, GnRH agonist triptorelin did not induce apoptosis. On the contrary, by activating NFκB, triptorelin even seems to protect against apoptosis [39][40].
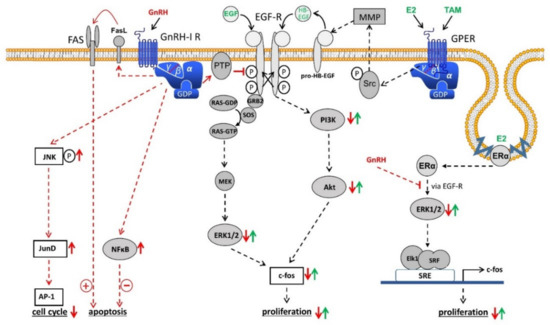
Figure 1. Schematic representation of GnRH-I receptor signaling in ovarian cancer cells. Binding of GnRH-I and GnRH-I analogs causes G-protein αi-mediated activation of a PTP, resulting in dephosphorylated EGF-R and inhibition of EGF-R signaling through ERK1/2 or PI3K/AKT pathway leading to reduction of proliferation. GnRH-I-induced activation of PTP also inhibits the signal transduction of GPER and membrane-associated ERα through transactivation of EGF-R. In addition, GnRH induces activation of NFκB to reduce and FAS-ligand to increase apoptosis. Finally, GnRH agonists activate the JNK/AP-1 pathway resulting in increased G0/1 phase of the cell cycle and decreased synthesis of DNA. Further details are described in the text. Abbreviations: Akt, protein kinase B (PKB); AP-1, activator protein-1; E2, estradiol; EGF, epidermal growth factor; EGF-R, EGF receptor; ERα, estrogen receptor α; ERK1/2, p44/42 mitogen-activated protein (MAP) kinase; FAS, Fas receptor; FasL, Fas ligand; GDP, guanosine diphosphate; GnRH, gonadotropin-releasing hormone; GPER, G-protein coupled estrogen receptor; GRB2, growth factor receptor-bound protein 2; GTP, guanosine triphosphate; HB-EGF, heparin-binding EGF-like growth factor; JNK, c-Jun N-terminal kinase; JunD, transcription factor JunD; MEK, mitogen-activated protein kinase kinase (MAP2K); MMP, matrix metalloproteinase; NFkB, nucleus factor kB; PI3K, phosphoinositide 3-kinase; PTP, protein tyrosine phosphatase; RAS, G-protein rat sarcoma; SOS, guanine nucleotide exchange factor “son of sevenless”; SRE, serum response element; Src, tyrosine kinase cellular sarcoma; SRF, serum response factor; TAM, tamoxifen.
In human cells from ovarian cancer, GnRH agonists act not only on the mitogenic signaling of growth factor receptors. They also induce the activation of activator protein-1 (AP-1). In addition, GnRH agonists promote activation of JNK, which triggers AP-1 [41]. In ovarian cancer cells, GnRH agonists stimulate neither phospholipase C (PLC) nor protein kinase C (PKC) [5]. In addition, GnRH agonists inhibit the mitogen-activated protein kinase (MAPK, ERK) activity induced by growth factors [5]. Therefore, GnRH-induced JNK/AP-1 activation depends not on the AP-1 activators PKC or MAPK (ERK).
Since the JNK/c-jun signal transduction is activated by antiproliferative GnRH agonists and since JNK/c-jun is integrated into the reduction of cell growth in different systems, it seems plausible that the JNK/c-jun signal transduction participates in the inhibiting effects of GnRH agonists. GnRH agonists have also been shown to induce the binding of JunD to DNA, resulting in a reduction of cell growth, as evidenced by an increased G0/1 phase of the cell cycle and reduced synthesis of DNA [42].
Expression of the immediate-early response gene c-fos is induced by 17β-estradiol (E2) in estrogen receptor α (ERα)-positive ovarian cancer cell lines [43][44][45][46][47][48][49][50]. Via MAPK-dependent phosphorylation of Elk-1 the serum response element (SRE) is activated [51][52]. Since GnRH agonists inhibit EGF-induced cell proliferation and expression of c-fos through Ras/MAPK, it was interesting to know whether activation of SRE and expression of c-fos induced by E2 in ERα-positive cancer cells is also influenced by GnRH agonists and whether GnRH inhibits E2-driven cell proliferation [3]. Resting ERα-positive/ERβ-positive cancer cell lines were stimulated to proliferate by treatment with E2. Simultaneous treatment with GnRH agonists prevented this effect in a time- and dose-dependent manner [53]. ERα-negative/ERβ-positive cell lines were not affected by treatment with E2. Furthermore, E2 activates serum response element (SRE) and expression of c-fos in ERα-positive / ERβ-positive cell lines. This was inhibited by GnRH agonists [53]. GnRH agonists had no influence on the activation of estrogen response element (ERE) caused by E2. Transcriptional activation of SRE by ERα is due to activation of the mitogen-activated protein kinase (MAPK) pathway. GnRH inhibits this signal transduction, resulting in a decrease in E2-induced SRE activation followed by a decrease in E2-mediated c-fos expression. This is causative for a reduction in E2-induced cell growth [53].
Activation of PTP by GnRH also inhibits Src/MMP/HB-EGF signaling via the G-protein βγ subunit of G-protein coupled estrogen receptor 1 (GPER1), a membrane-bound estrogen receptor [54][55][56][57]. Due to the inhibition of GPER signaling, cancer cell proliferation by E2 in breast cancer cells without expression of ERα was inhibited [54][55][56]. In human breast cancer cells, GnRH analogs counteract EGF-dependent proliferation. GnRH analogs likely disrupt the change in growth regulation from estrogen dependency to EGF dependence that occurs after the acquisition of secondary resistance against 4OH-tamoxifen. This disruption of EGF receptor signal transduction re-sensitized the resistant cell lines for a therapeutical use of 4OH-Tamoxifen [58]. Especially in breast cancer, GnRH is not only involved in cancer cell proliferation. In metastatic breast cancer cells, GnRH inhibits cell invasiveness in vitro and metastasis in vivo [59][60][61][62][63]. In addition, it was shown that GnRH is involved in epithelial to mesenchymal transition (EMT) [63]. However, these effects of GnRH still have to be researched in ovarian cancer in the future.
References
- Langdon, S.; Crew, A.; Ritchie, A.; Muir, M.; Wakeling, A.; Smyth, J.; Miller, W. Growth inhibition of oestrogen receptor-positive human ovarian carcinoma by anti-oestrogens in vitro and in a xenograft model. Eur. J. Cancer 1994, 30, 682–686.
- Langdon, S.; Hirst, G.; Miller, E.; Hawkins, R.; Tesdale, A.; Smyth, J.; Miller, W. The regulation of growth and protein expression by estrogen in vitro: A study of 8 human ovarian carcinoma cell lines. J. Steroid Biochem. Mol. Biol. 1994, 50, 131–135.
- Gründker, C.; Günthert, A.R.; Westphalen, S.; Emons, G. Biology of the gonadotropin-releasing hormone system in gynecological cancers. Eur. J. Endocrinol. 2002, 146, 1–14.
- Emons, G.; Grundker, C.; Gunthert, A.R.; Westphalen, S.; Kavanagh, J.; Verschraegen, C. GnRH antagonists in the treatment of gynecological and breast cancers. Endocr. Relat. Cancer 2003, 10, 291–299.
- Dondi, D.; Limonta, P.; Moretti, R.M.; Marelli, M.M.; Garattini, E.; Motta, M. Antiproliferative effects of lu-teinizing hormone-releasing hormone (LHRH) agonists on human androgen-independent prostate cancer cell line DU 145: Evidence for an autocrine-inhibitory LHRH loop. Cancer Res. 1994, 54, 4091–4095.
- Limonta, P.; Dondi, D.; Moretti, R.M.; Maggi, R.; Motta, M. Antiproliferative effects of luteinizing hor-mone-releasing hormone agonists on the human prostatic cancer cell line LNCaP. J. Clin. Endocrinol. Metab. 1992, 75, 207–212.
- Limonta, P.; Moretti, R.M.; Dondi, D.; Marelli, M.M.; Motta, M. Androgen-dependent prostatic tumors: Bio-synthesis and possible actions of LHRH. J. Steroid Biochem. Mol. Biol. 1994, 49, 347–350.
- Cheng, K.W.; Leung, P.C. The expression, regulation and signal transduction pathways of the mammalian gonadotropin-releasing hormone receptor. Can. J. Physiol. Pharm. 2000, 78, 1029–1052.
- Naor, Z. Signal Transduction Mechanisms of Ca 2+ Mobilizing Hormones: The Case of Gonadotropin-Releasing Hormone. Endocr. Rev. 1990, 11, 326–353.
- Kraus, S.; Naor, Z.; Seger, R. Intracellular Signaling Pathways Mediated by the Gonadotropin-Releasing Hormone (GnRH) Receptor. Arch. Med. Res. 2001, 32, 499–509.
- McArdle, C.A.; Franklin, J.; Green, L.; Hislop, J.N. Signalling, cycling and desensitisation of gonadotrophin-releasing hormone receptors. J. Endocrinol. 2002, 173, 1–11.
- Ruf, F.; Fink, M.Y.; Sealfon, S.C. Structure of the GnRH receptor-stimulated signaling network: Insights from genomics. Front. Neuroendocr. 2003, 24, 181–199.
- Berridge, M.J. Inositol trisphosphate and calcium signalling. Nat. Cell Biol. 1993, 361, 315–325.
- Keizer, J.; Li, Y.X.; Stojilković, S.; Rinzel, J. InsP3-induced Ca2+ excitability of the endoplasmic reticulum. Mol. Biol. Cell 1995, 6, 945–951.
- Harris, D.; Bonfil, D.; Chuderland, D.; Kraus, S.; Seger, R.; Naor, Z. Activation of MAPK Cascades by GnRH: ERK and Jun N-Terminal Kinase Are Involved in Basal and GnRH-Stimulated Activity of the Glycoprotein Hormone LHβ-Subunit Promoter. Endocrinology 2002, 143, 1018–1025.
- Zhang, T.; Roberson, M.S. Role of MAP kinase phosphatases in GnRH-dependent activation of MAP kinases. J. Mol. Endocrinol. 2006, 36, 41–50.
- Bonfil, D.; Chuderland, D.; Kraus, S.; Shahbazian, D.; Friedberg, I.; Seger, R.; Naor, Z. Extracellular Signal-Regulated Kinase, Jun N-Terminal Kinase, p38, and c-Src Are Involved in Gonadotropin-Releasing Hormone-Stimulated Activity of the Glycoprotein Hormone Follicle-Stimulating Hormone β-Subunit Promoter. Endocrinology 2004, 145, 2228–2244.
- Levi, N.L.; Hanoch, T.; Benard, O.; Rozenblat, M.; Harris, D.; Reiss, N.; Naor, Z.; Seger, R. Stimulation of Jun N-Terminal Kinase (JNK) by Gonadotropin-Releasing Hormone in Pituitary αT3–1 Cell Line Is Mediated by Protein Kinase C, c-Src, and CDC42. Mol. Endocrinol. 1998, 12, 815–824.
- Roberson, M.S.; Zhang, T.; Li, H.L.; Mulvaney, J.M. Activation of the p38 Mitogen-Activated Protein Kinase Pathway by Gonadotropin-Releasing Hormone. Endocrinology 1999, 140, 1310–1318.
- Gründker, C.; Völker, P.; Emons, G. Antiproliferative signaling of luteinizing hormone-releasing hormone in human endometrial and ovarian cancer cells through G protein alpha(I)-mediated activation of phosphotyrosine phosphatase. Endocrinology 2001, 142, 2369–2380.
- Limonta, P.; Moretti, R.M.; Marelli, M.M.; Dondi, D.; Parenti, M.; Motta, M. The luteinizing hormone-releasing hormone receptor in human prostate cancer cells: Messenger ribonucleic acid expression, molecular size, and signal transduction pathway. Endocrinology 1999, 140, 5250–5256.
- Dobkin-Bekman, M.; Naidich, M.; Pawson, A.J.; Millar, R.P.; Seger, R.; Naor, Z. Activation of mitogen-activated protein kinase (MAPK) by GnRH is cell-context dependent. Mol. Cell Endocrinol. 2006, 252, 184–190.
- Lee, M.T.; Liebow, C.; Kamer, A.R.; Schally, A.V. Effects of epidermal growth factor and analogues of lute-inizing hormone-releasing hormone and somatostatin on phosphorylation and dephosphorylation of tyro-sine residues of specific protein substrates in various tumors. Proc. Natl. Acad. Sci. USA 1991, 88, 1656–1660.
- Furui, T.; Imai, A.; Takagi, H.; Horibe, S.; Fuseya, T.; Tamaya, T. Phosphotyrosine Phosphatase-Activity in Membranes from Endometrial Carcinoma. Oncol. Rep. 1995, 2, 1055–1057.
- Imai, A.; Takagi, H.; Horibe, S.; Fuseya, T.; Tamaya, T. Coupling of gonadotropin-releasing hormone recep-tor to Gi protein in human reproductive tract tumors. J. Clin. Endocrinol. Metab. 1996, 81, 3249–3253.
- Imai, A.; Takagi, H.; Furui, T.; Horibe, S.; Fuseya, T.; Tamaya, T. Evidence for coupling of phosphotyrosine phosphatase to gonadotropin-releasing hormone receptor in ovarian carcinoma membrane. Cancer 1996, 77, 132–137.
- Gründker, C.; Völker, P.; Schulz, K.-D.; Emons, G. Luteinizing Hormone—Releasing Hormone Agonist Triptorelin and Antagonist Cetrorelix Inhibit EGF-Induced c-fos Expression in Human Gynecological Cancers. Gynecol. Oncol. 2000, 78, 194–202.
- Miller, W.R.; Scott, W.N.; Morris, R.; Fraser, H.M.; Sharpe, R.M. Growth of human breast cancer cells inhib-ited by a luteinizing hormone-releasing hormone agonist. Nature 1985, 313, 231–233.
- Yano, T.; Pinski, J.; Halmos, G.; Szepesházi, K.; Groot, K.; Schally, A.V. Inhibition of growth of OV-1063 human epithelial ovarian cancer xenografts in nude mice by treatment with luteinizing hormone-releasing hormone antagonist SB-75. Proc. Natl. Acad. Sci. 1994, 91, 7090–7094.
- Moretti, R.M.; Marelli, M.M.; Dondi, D.; Poletti, A.; Martini, L.; Motta, M.; Limonta, P. Luteinizing hormone-releasing hormone agonists interfere with the stimulatory actions of epidermal growth factor in human prostatic cancer cell lines, LNCaP and DU 145. J. Clin. Endocrinol. Metab. 1996, 81, 3930–3937.
- Shirahige, Y.; Cook, C.; Pinski, J.; Halmos, G.; Nair, R.; Schally, A. Treatment with Luteinizing-Hormone-Releasing Hormone Antagonist sb-75 Decreases Levels of Epidermal Growth-Factor Receptor and its Messenger-RNA in ov-1063 Human Epithelial Ovarian-Cancer Xenografts in Nude-Mice. Int. J. Oncol. 1994, 5, 1031–1035.
- Kéri, G.; Balogh, Á.; Szöke, B.; Teplán, I.; Csuka, O. Gonadotropin-Releasing Hormone Analogues Inhibit Cell Proliferation and Activate Signal Transduction Pathways in MDA-MB-231 Human Breast Cancer Cell Line. Tumor Biol. 1991, 12, 61–67.
- Liebow, C.; Lee, M.T.; Kamer, A.R.; Schally, A.V. Regulation of luteinizing hormone-releasing hormone re-ceptor binding by heterologous and autologous receptor-stimulated tyrosine phosphorylation. Proc. Natl. Acad. Sci. USA 1991, 88, 2244–2248.
- Hershkovitz, E.; Marbach, M.; Bosin, E.; Levy, J.; Roberts, C.T., Jr.; LeRoith, D.; Schally, A.V.; Sharoni, Y. Lu-teinizing hormone-releasing hormone antagonists interfere with autocrine and paracrine growth stimula-tion of MCF-7 mammary cancer cells by insulin-like growth factors. J. Clin. Endocrinol. Metab. 1993, 77, 963–968.
- Fuseya, T.; Imai, A.; Horibe, S.; Takagi, A.; Tamaya, T. Evidence for common signalling pathways of GnRH receptor and Fas in tumors. Oncol. Rep. 1996, 3, 1111–1113.
- Imai, A.; Horibe, S.; Takagi, A.; Ohno, T.; Tamaya, T. Frequent expression of Fas in gonadotropin-releasing hormone receptor-bearing tumors. Eur. J. Obstet. Gynecol. Reprod. Biol. 1997, 74, 73–78.
- Imai, A.; Takagi, A.; Horibe, S.; Takagi, H.; Tamaya, T. Fas and Fas ligand system may mediate antiproliferative activity of gonadotropin-releasing hormone receptor in endometrial cancer cells. Int. J. Oncol. 1998, 13, 97–197.
- Imai, A.; Takagi, A.; Horibe, S.; Takagi, H.; Tamaya, T. Evidence for Tight Coupling of Gonadotropin-Releasing Hormone Receptor to Stimulated Fas Ligand Expression in Reproductive Tract Tumors: Possible Mechanism for Hormonal Control of Apoptotic Cell Death 1. J. Clin. Endocrinol. Metab. 1998, 83, 427–431.
- Gunthert, A.R.; Grundker, C.; Bottcher, B.; Emons, G. Luteinizing hormone-releasing hormone (LHRH) in-hibits apoptosis induced by cytotoxic agent and UV-light but not apoptosis mediated through CD95 in hu-man ovarian and endometrial cancer cells. Anticancer Res. 2004, 24, 1727–1732.
- Gründker, C.; Schulz, K.; Günthert, A.R.; Emons, G. Luteinizing Hormone-Releasing Hormone Induces Nuclear Factorκ B-Activation and Inhibits Apoptosis in Ovarian Cancer Cells. J. Clin. Endocrinol. Metab. 2000, 85, 3815–3820.
- Grundker, C.; Schlotawa, L.; Viereck, V.; Emons, G. Protein kinase C-independent stimulation of activator protein-1 and c-Jun N-terminal kinase activity in human endometrial cancer cells by the LHRH agonist triptorelin. Eur. J. Endocrinol. 2001, 145, 651–658.
- Günthert, A.R.; Gründker, C.; Hollmann, K.; Emons, G. Luteinizing hormone-releasing hormone induces JunD—DNA binding and extends cell cycle in human ovarian cancer cells. Biochem. Biophys. Res. Commun. 2002, 294, 11–15.
- Bonapace, I.M.; Addeo, R.; Altucci, L.; Cicatiello, L.; Bifulco, M.; Laezza, C.; Salzano, S.; Sica, V.; Bresciani, F.; Weisz, A. 17 beta-Estradiol overcomes a G1 block induced by HMG-CoA reductase inhibitors and fosters cell cycle progression without inducing ERK-1 and -2 MAP kinases activation. Oncogene 1996, 12, 753–763.
- Doucas, V.; Spyrou, G.; Yaniv, M. Unregulated expression of c-Jun or c-Fos proteins but not Jun D inhibits oestrogen receptor activity in human breast cancer derived cells. EMBO J. 1991, 10, 2237–2245.
- Duan, R.; Porter, W.; Safe, S. Estrogen-Induced c-fos Protooncogene Expression in MCF-7 Human Breast Cancer Cells: Role of Estrogen Receptor Sp1 Complex Formation. Endocrinology 1998, 139, 1981–1990.
- Van Der Burg, B.; De Groot, R.P.; Isbrücker, L.; Kruijer, W.; De Laat, S.W. Stimulation of TPA-Responsive Element Activity by a Cooperative Action of Insulin and Estrogen in Human Breast Cancer Cells. Mol. Endocrinol. 1990, 4, 1720–1726.
- Van der Burg, B.; de Groot, R.P.; Isbrucker, L.; Kruijer, W.; de Laat, S.W. Oestrogen directly stimulates growth factor signal transduction pathways in human breast cancer cells. J. Steroid Biochem. Mol. Biol. 1991, 40, 215–221.
- Van Der Burg, B.; Van Selm-Miltenburg, A.J.; De Laat, S.W.; Van Zoelen, E.J. Direct effects of estrogen on c-fos and c-myc protooncogene expression and cellular proliferation in human breast cancer cells. Mol. Cell. Endocrinol. 1989, 64, 223–228.
- Weisz, A.; Bresciani, F. Estrogen regulation of proto-oncogenes coding for nuclear proteins. Crit. Rev. Oncog. 1993, 4, 361–388.
- Wilding, G.; Lippman, M.E.; Gelmann, E.P. Effects of steroid hormones and peptide growth factors on pro-tooncogene c-fos expression in human breast cancer cells. Cancer Res. 1988, 48, 802–805.
- Duan, R.; Xie, W.; Burghardt, R.C.; Safe, S. Estrogen Receptor-mediated Activation of the Serum Response Element in MCF-7 Cells through MAPK-dependent Phosphorylation of Elk-1. J. Biol. Chem. 2001, 276, 11590–11598.
- Duan, R.; Xie, W.; Li, X.; McDougal, A.; Safe, S. Estrogen regulation of c-fos gene expression through phosphatidylinositol-3-kinase-dependent activation of serum response factor in MCF-7 breast cancer cells. Biochem. Biophys. Res. Commun. 2002, 294, 384–394.
- Grundker, C.; Gunthert, A.R.; Hellriegel, M.; Emons, G. Gonadotropin-releasing hormone (GnRH) agonist triptorelin inhibits estradiol-induced serum response element (SRE) activation and c-fos expression in hu-man endometrial, ovarian and breast cancer cells. Eur. J. Endocrinol. 2004, 151, 619–628.
- Girgert, R.; Emons, G.; Gründker, C. Inactivation of GPR30 reduces growth of triple-negative breast cancer cells: Possible application in targeted therapy. Breast Cancer Res. Treat. 2012, 134, 199–205.
- Girgert, R.; Emons, G.; Gründker, C. Inhibition of GPR30 by estriol prevents growth stimulation of triple-negative breast cancer cells by 17β-estradiol. BMC Cancer 2014, 14, 935.
- Girgert, R.; Emons, G.; Gründker, C. 17β-estradiol-induced growth of triple-negative breast cancer cells is prevented by the reduction of GPER expression after treatment with gefitinib. Oncol. Rep. 2016, 37, 1212–1218.
- Luttrell, L.M.; Daaka, Y.; Lefkowitz, R.J. Regulation of tyrosine kinase cascades by G-protein-coupled recep-tors. Curr. Opin. Cell Biol. 1999, 11, 177–183.
- Günthert, A.R.; Gründker, C.; Olota, A.; Läsche, J.; Eicke, N.; Emons, G. Analogs of GnRH-I and GnRH-II inhibit epidermal growth factor-induced signal transduction and resensitize resistant human breast cancer cells to 4OH-tamoxifen. Eur. J. Endocrinol. 2005, 153, 613–625.
- Von Alten, J.; Fister, S.; Schulz, H.; Viereck, V.; Frosch, K.-H.; Emons, G.; Gründker, C. GnRH analogs reduce invasiveness of human breast cancer cells. Breast Cancer Res. Treat. 2006, 100, 13–21.
- Ziegler, E.; Hansen, M.-T.; Haase, M.; Emons, G.; Gründker, C. Generation of MCF-7 cells with aggressive metastatic potential in vitro and in vivo. Breast Cancer Res. Treat. 2014, 148, 269–277.
- Olbrich, T.; Ziegler, E.; Türk, G.; Schubert, A.; Emons, G.; Gründker, C. Kisspeptin-10 inhibits bone-directed migration of GPR54-positive breast cancer cells: Evidence for a dose-window effect. Gynecol. Oncol. 2010, 119, 571–578.
- Magliocco, A.; Egan, C. Breast Cancer Metastasis: Advances Trough the Use of In Vitro Co-Culture Model Systems. In Breast Cancer-Focusing Tumor Micriinvironment, Stem Cells and Metastasis; Gunduz, M., Gunduz, E., Eds.; InTech: Rijeka, Croatia, 2011; Volume 1.
- Hellinger, J.W.; Hüchel, S.; Goetz, L.; Bauerschmitz, G.; Emons, G.; Gründker, C. Inhibition of CYR61-S100A4 Axis Limits Breast Cancer Invasion. Front. Oncol. 2019, 9, 1074.

