 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Vyacheslav Kosorukov | + 3596 word(s) | 3596 | 2020-10-15 12:00:49 | | | |
| 2 | Conner Chen | + 1 word(s) | 3597 | 2021-04-01 15:45:44 | | | | |
| 3 | Conner Chen | + 1 word(s) | 3597 | 2021-04-01 15:46:12 | | | | |
| 4 | Conner Chen | + 217 word(s) | 3814 | 2021-04-08 03:32:55 | | |
Video Upload Options
This entry provides an overview of currently available approaches applied for neoantigens discovery—tumor-specific peptides that appeared due to the mutation process and distinguish tumors from normal tissues. Focusing on genomics-based approaches and computational pipelines, we cover all steps required for selecting appropriate candidate peptides starting from NGS-derived data. Moreover, additional approaches such as mass-spectrometry-based and structure-based methods are discussed highlighting their advantages and disadvantages. This review also provides a description of available complex bioinformatics pipelines ensuring automated data processing resulting in a list of neoantigens. We propose the possible ideal pipeline that could be implemented in the neoantigens identification process. We discuss the integration of results generated by di erent approaches to improve the accuracy of neoantigens selection.
1. Genomics-Based Approaches and Current Bioinformatics Pipelines
Currently, genomics-based strategies are some of the most promising in the field of neoantigen development. The widespread use of NGS-based techniques stimulates the development of bioinformatics tools, including those that are implemented in clinical practice. In the context of neoantigen discovery, it is important to note that the accuracy of peptide selection significantly depends on bioinformatics pipelines that are applied to the processing of the data obtained by WES and RNA-seq. A limited number of complex pipelines for these purposes were developed and described during the last several years [1][2][3][4][5]. For detailed information of selected currently available genomics-centered pipelines. Most of them combine principally the same set of tools (in terms of tool class) intended to carry out the main steps of analysis including raw data pre-processing to remove low-quality data, mapping to the reference genome, somatic mutation calling, mutated peptide sequences isolation and peptide ranking according to their predicted capacity to be presented on the cell surface by the MHC and recognized by the TCR. A general overview of these steps is shown in Figure 1. Current comprehensive best practices in bioinformatics in the context of neoantigen identification are presented in [6]. In this report, authors not only describe the appropriate tools for each analysis step but also provide fundamental guidelines that could serve as a basis for creating standardized consensus rules for neoantigen research.
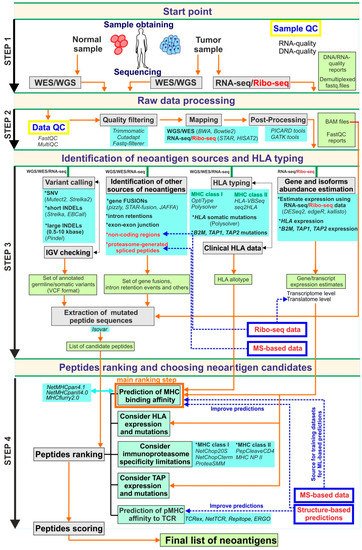
Figure 1. The schematic description of the possible ideal genomics-centric pipeline for neoantigen identification. In this scheme, the pipeline is formally split into four steps. Step 1 is related to sample obtaining, DNA/RNA-extraction, libraries preparation and high-throughput sequencing. Step 2 is associated with raw NGS data processing, quality-filtering and obtaining aligned to reference reads in an appropriate format suitable for downstream analysis. The aim of Step 3 is to obtain all possible information from processed NGS including all variants set, HLA allotype, expression estimations, as well as, candidate peptide sequences that further to prioritization procedures of Step 4. At the final Step 4 candidate peptide list obtaining based on identified variants used for peptide ranking using mainly MHC binding estimators. Additional options such as TCR-pMHC binding affinity scoring, TAP-transport, etc., should be considered during the prioritization step. Grey boxes are reflected main procedures that should be done in each step; light green boxes contain data that should be generated on each step; light blue boxes present possible tools that could be applied for the corresponding procedure; blue boxes with red text (as well as red text alone) are related to additional approaches that could add value to this pipeline; the orange arrows show the information flow through pipeline workflow; the blue dashed arrows highlight the steps that could be improved by additional approaches.
In general, the preliminary data that can be extracted during WES/WGS and RNA-seq analysis consist of bam-files containing sequenced reads aligned to the reference genome, a set of germline/somatic mutations, the patient’s HLA allotypes, estimates of gene expression levels as well as information regarding the abundance of transcript isoforms. Somatic mutation data and RNA-seq alignments are also used to determine mutant protein sequences.
Somatic variant identification is one of the most important and, at the same time, one of the most delicate parts of all pipelines. It is now firmly established that not only neoantigens arising from single nucleotide variants (SNV) could be candidates for vaccines. Other mutation types are also considered to be sources of neoantigens. Among them are INDELs (short insertions and deletions) [7], gene fusions [8], exon-exon junctions [9], intron retentions [10] and some other alternative splicing events [11]. RNA transcription and splicing errors [12], as well as RNA editing examples [11], could also be recognized as neoantigen sources. Non-coding genome regions such as non-coding exons, UTRs, non-coding RNAs, and others could also be neoantigen sources [13]. This list could potentially be extended by V(D)J recombination and somatic hypermutation events [14][15] that are important for blood malignancies, and sequences of viruses that are associated with some tumors [16][17]. Proteasome-generated spliced peptides, as well as peptides bearing tumor-specific post-translational modifications, could also be a source of neoantigens [18][19], but they are out of the scope of NGS-based approaches. It was reported that neoantigens resulting from non-SNV variants could make up to 15% of all neoantigens [20]. Some authors state that non-coding regions could be the main source of neoepitopes [13]. Moreover, recent proteogenomics studies of ovarian cancer revealed that the composition of tumor-specific antigens resulting from non-mutated non-exonic regions includes 29% of intronic and 22% of intergenic sequences, and most importantly, many of them are shared across tumors [21]. Thus the variety of mutation types makes it necessary to select the right tool for the identification of each type if this tool is available. Tools for the identification of some of the mutation types listed above are discussed in [6]; additionally, comprehensive comparisons can be found in [22][23][24]. Mutect2 and Strelka2 are the most reliable somatic variant callers for SNV identification [22]. It is advisable to run several somatic callers simultaneously, which could potentially improve calling accuracy [25]. It is also good practice to conduct manual verification of somatic mutation caller results by viewing them in genomic browsers and to carry out additional validation utilizing targeted sequencing approaches [6]. Identification of other neoantigen sources is also possible due to tools such as Strelka [26] and EBCall [27], which are designed for INDELs calling, and Pindel [28], which is a specialized tool for calling large INDELs. A variety of tools for gene fusion identification were also developed, such as INTEGRATE [29] (and INTEGRATE-neo pipeline [30]), STAR-fusion [31], etc. There is now a clear demand for the development of tools that can provide proper identification of all the neoantigen sources listed above.
Furthermore, after detecting all the variants of interest, one wants to know whether they could, in principle, yield a neoantigen that has a chance to bind to the MHC molecule. Firstly, it is well known that the immunoproteasome has a limited specificity, which means that not every possible mutated peptide will be produced during protein degradation [32]. Secondly, not all peptides produced by the proteasome would reach the required compartment of cells and could, in principle, interact with the MHC. It is known that before being presented by MHC class I, peptides are at first transported into the EPR (endoplasmic reticulum) by special transporters known as TAP (Transporter associated with antigen processing) and then trimmed by ER-related aminopeptidases (ERAP) [33]. There are several tools assessing TAP transport efficiency for peptides [34][35][36][37] and a number of tools that allow us to take proteasome cleavage specificity into account, such as NetChop20S, ProteaSMM [36][38] for MHC class I pathway and PepCleaveCD4, MHC II NP [39][40] for MHC class II pathway. It should also be taken into consideration that genes that code transporters of antigen-presenting machinery such as TAP1, TAP2, B2M, etc., can have mutations influencing their activity, and that these genes can have different expression levels in various tumor types, which has an additional impact on peptide presentation [41][42]. Thus, taking proteasome cleavage specificity and TAP transport limitations into account, the final list of peptides based on identified somatic variants should be created and subjected to subsequent prioritization procedures.
As mentioned above, currently available epitope prediction algorithms are based on the idea that the affinity of the peptide to a given MHC class molecule is the dominant contributor to neoantigen immunogenicity, and thus this parameter is considered to be the primary factor for peptide prioritization. It relies on the observation that only about 1 of 10,000 peptides resulting from protein degradation will be presented by the MHC [43]. It is also well-known that different MHC allotypes differ in specificity with respect to peptide binding. Therefore, it is crucial to know the HLA type before ranking peptides. The gold standard for HLA allotype determination is clinical HLA typing by sequence-specific PCR [44][45]. However, currently available HLA typers based on WES/RNA-seq data provide a high enough accuracy rate and can also be used for HLA allotype identification when a clinical HLA type is unavailable. Although HLA class I typing algorithms can reach an accuracy of up to 99% [46][47], HLA class II typers remain less effective and require additional development. It is no less important to estimate HLA locus gene expression as well as to determine somatic mutation patterns in this locus, as they both can be a cause of neoantigen presentation loss leading to resistance to immunotherapy [48][49][50].
Prediction of peptide-MHC binding affinity is the most critical step of the neoantigen discovery process. Many tools for such analysis exist [51][52][53][54]. These tools utilize large-scale peptide-MHC binding affinity data derived from biochemical measurements and eluted ligands data obtained by high-throughput mass-spectrometry analysis of MHC ligandome [51][55] to train machine learning-based classifiers that can identify binders and non-binders and calculate affinity scores. The machine learning approaches include linear regression (LR) and artificial neural networks (ANN). Depending on the experimental data that are used to train these algorithms, they can be classified on binding affinity (BA) trained methods, eluted ligands (EL) trained methods, and mixed trained methods utilizing both BA- and EL-datasets. Since the performance of different algorithms varies, a number of comprehensive benchmarking studies were carried out to compare the accuracy of these tools [56][57][58][59]. For instance, according to [57], where a dataset for 32 HLA class I and 24 HLA class II was used, ANN-based approaches showed better performance than LR-based, and among 19 predictors that were benchmarked, MHCflurry (AUC = 0.911 ± 0.010) and ann_align (AUC = 0.911 ± 0.004) showed the highest accuracy in terms of the AUC (Area Under ROC Curve) for MHC class I 9-mer and MHC class II 15-mer, respectively, in binding versus non-binding classification. In another benchmarking study [56], using an experimentally validated dataset with binding affinity data for 743 peptides (8- to 11-mers), derived from the HPV16 E6 and E7 proteins, none of the algorithms outperformed the others. However, different algorithms showed better performance for particular HLA types and peptide lengths [56]. In one of the most recent benchmarking studies [60], the performance of 15 algorithms was tested on a dataset described in [61], which contains 220 naturally processed vaccinia virus (VACV) peptides that were eluted from VACV-infected cells and tested for T cell immune response in infected C57Bl/6 mice. ANN-based NetMHCpan 4.0-L (AUC = 0.977), NetMHCpan 4.0-B (AUC = 0.975) and MHCflurry-L (AUC = 0.973) were reported to achieve the best performance which was in general agreement with the results previously reported in [57]. More recently, improved versions of NetMHCpan (v.4.1) and NetMHCIIpan (v.4.0) as well as MHCflurry (v.2.0) were presented [51][53]. In [51] NNAlign_MA was used to update NetMHCpan and NetMHCIIpan which outperformed the current state-of-the-art methods including NetMHCpan 4.0 and MHCflurry. O’Donnell et al. incorporated an antigen processing predictor that uses data on MHC ligands, identified by mass-spectrometry, into MHCflurry 2.0 [53], allowing it to achieve better accuracy than the currently available tools. It seems logical that the simultaneous use of several MHC-binding predictors could improve peptide prioritization. It should be noted that currently available MHC-binding predictors suffer from inadequate support for rare MHC alleles and poor performance for MHC class II molecules. Another significant inherited weakness of this approach is the failure to consider the effect of post-translational modification on binding affinity. Despite these weaknesses, this approach is the gold standard in the prediction of MHC-peptide interactions.
It is well-known that not all peptides presented by the MHC (pMHC complexes) trigger T cell activation [62][63]. For instance, in [63], the authors summarized data on candidate neoantigens predicted to be MHC-binders from 13 suitable published works, which included information about assessing the peptides’ immunogenic potential. It turned out that only 53 of 1948 neopeptide-MHC combinations elicited T cell response. In [64] it was reported that among 50 long peptides (MHC-binding prediction was performed using NetMHC 3.0) that were selected based on non-synonymous 563 somatic mutations in genes that are expressed in B16F10 murine melanoma, only one-third were immunogenic, and 60% of them elicited immune response directed against the mutated sequences. According to [65], only 25 of 66 27-mer peptides selected by predicted binding affinity to MHC I and MHC II and expression level were immunogenic according to IFNg ELISpot assay. Remarkably, in mouse models, the majority of immunogenic neoantigens (up to 90%) were associated with CD4+ T cell response [64][65][66]. Since the primary goal of neoantigen identification (in the context of cancer vaccines development) is to select those that would trigger or boost T-cell-mediated immune response (preferably CD8+ T cell response), it is essential to know which of the peptides with a high MHC binding affinity will be recognized by T-cells. This brings about the challenge of determining the specificity of MHC-epitope-TCR interactions, which could be an additional layer of the neoantigens ranking process. It is an established fact that T cells recognize pMHC complexes predominantly by the complementarity determining region 3 (CDR3) loops of the TCR [67]. Based on the fact that different individuals having different TCR repertoires can recognize the same epitopes arising from the same agents (e.g., immunodominant viral epitopes [68][69][70]), one may suggest that such epitopes have intrinsic patterns that make them more recognizable by the TCR. On the other hand, it was observed that TCR repertoires that are specific to the same epitope have similarities in their core sequences [71]. Such reasoning allows us to suggest that it is possible to perform a simulation based on sequences of peptides and TCR repertoires. Several approaches to predicting epitope-TCR binding were developed (e.g., TCRex [72], NetTCR [73], Repitope [74], ERGO [75], Deepwalk approach [76]). For instance, TCRex is based on the principle that similar TCR sequences often target the same epitope [72], Repitope is based on the idea that sequences of epitopes contain some intrinsic hidden pattern that is prone to activating T cell response [74]. Unfortunately, this class of tools is at the initial stage of development, and their prediction power suffers from insufficient training data on TCR–epitope interactions. Meanwhile, in the present time, other strategies are being successfully implemented to improve the immunogenicity of neoantigens [77][78]. Thus, in [77] the weak B16F10 neoantigens described in [64] were fused to the transmembrane domain of diphtheria toxin (DTT), significantly enhancing their ability to elicit CD8+ T cell response and inhibit tumor growth. A bi-adjuvant vaccine containing a neoantigen supplemented with two adjuvants such as the Toll-like receptor (TLR) 7/8 agonist R848 and the TLR9 agonist CpG, boosted the immunogenicity of the neoantigen due to efficient co-delivery and synergism of adjuvants [78].
2. Mass Spectrometry-Based Approaches
Genomics-based approaches represent the gold standard that is applied for neoantigen vaccine development, including in silico peptide prediction. Neoantigen candidate selection relies on the spectra of somatic mutations identified by WES/RNA-seq. This approach suffers from a lack of direct experimental evidence of the real presence of predicted epitopes on the cell surface as a complex with MHC molecules [79][80]. Lacking data could be obtained using high-throughput mass spectrometry techniques [79][80] that at present allow us to analyze large amounts of peptides or whole proteins simultaneously. This review does not aim to give a detailed characterization of MS-based approaches; for a comprehensive review on this topic, the reader could refer to [81][79][80].
A typical MS workflow (IP-based) starts with immunoprecipitation of MHC-peptide complexes using beads conjugated with MHC-specific antibodies or beads bound with dummy antibodies as negative controls. Subsequent washing steps ensure the removal of unbound and non-specifically bound peptides, whereupon the eluted material is subjected to MS analysis. Another strategy is mild acid elution (MAE) of MHC-bound peptides from the cell surface by treatment under mildly acidic conditions [82], followed by MS analysis. This method has a significant false-positive rate and low specificity due to contamination with a large quantity of non-specific peptides. Detailed comparison of IP- and MAE-based approaches are presented in [83]. To find information on MHC peptidome identification by MS approach, the reader could refer to Zhang et al. [80] where authors provide a summary of 40 studies that were carried out from 1990 to 2019.
Unlike the genomics-based approach, which only provides for neoantigen prediction, mass-spectrometry allows us to take a real snapshot of the total MHC-bound protein interactome. Additionally, it could reveal not only neoantigens that originate from somatic mutation variants but also those which arise due to proteasome-mediated peptide splicing [84][85]. Using mass-spectrometry, it was shown that the proportion of spliced peptides relative to peptides displayed by HLA class I varies from 2-6% reported in [86] to 30% reported in [18]. Moreover, MS allows us to identify the post-translational modifications (PTM) of peptides bound to the MHC, thus shedding light on the importance of PTM for binding affinity [87]. Mass-spectrometry derived data served for the development of the first tool allowing to predict the interaction between HLA class I molecules and phosphorylated peptides [88]. In addition, MS-based profiling of HLA peptidome could generate high-quality training data that could potentially significantly improve current prediction models [51][55][89], and could also be used for benchmarking available tools.
Nevertheless, MS also has some limitations. They include low sensitivity and reproducibility. These problems are especially acute for low-abundance peptides, including tumor-specific neoantigens. Moreover, the washing stages of MHC-peptide complexes during IP could result in a loss of bound peptides. These issues impose a limitation on the initial quantities of biological material. For typical experiments, 1 g of tumor tissue or anywhere from a hundred million to billions of cells are required [83]. It should also be noted that cancer cells and tumor tissues have different HLA molecules; thus, peptides that were identified from this type of material are relevant for different HLA molecules, adding the problem of specificity of the HLA ligandome.
In summary, by combining genomics-based predictions with high-throughput HLA-ligandome mass-spectrometry data, the performance of neoantigen discovery procedures could be significantly enhanced. For instance, the currently available ProGeo-neo pipeline [90] utilizes LC-MS/MS data to verify NGS-based derived neoantigen candidates.
3. Structure-Based Approaches
Structure-based predictions are another option that can improve the state of the art in the context of neoantigen discovery [91][92]. While the genomics-based approach utilizes sequence-based methods, the structure-based prediction is additionally capable of uncovering the significance of peptide structure and physicochemical properties, as well as the importance of post-translational modifications, such as phosphorylation [93], citrullination [94], and glycosylation [95], for peptide binding to the MHC and the TCR. Moreover, structure-based approaches could yield predictions that will be applicable to all types of MHC and TCR receptors, mitigating the limitations of small training datasets for rare MHC alleles, which are required for machine learning-based predictions.
Despite the slow progress in the development of structure-based approaches due to the need for serious computational resources and high-resolution models, some attempts in this direction were made. In 2000 Schueler-Furman et al. [96] developed an approach utilizing a pairwise potential matrix that can be applied to a wide range of MHC I molecules for predicting peptide binding. In the following years, new algorithms for the prediction of peptide-MHC complexes binding were developed. PePSSI (peptide-MHC prediction of structure through solvated interfaces) [97] is an approach that allows predicting the structure of peptides bound to HLA-A2. It includes a sampling of peptide backbone conformations and flexible movement of MHC side chains and can explicitly take water molecules at the pMHC interface into account. Initially, PePSSI was tested to predict the conformation of eight peptides bound to HLA-A2, for which crystallography data are available. Analysis of predicted structures in comparison with structures derived from X-ray models showed them to be in good agreement. In [98] a method based on molecular dynamics simulations and estimation of free energy of binding between peptides and HLA molecules was proposed. Another approach, HLAffy, is based on the strength of a mechanistic model of peptide-HLA recognition [99]. It can predict epitopes for any class I HLA by assessing the binging affinity of peptide-HLA complexes by learning pair potentials that are important for peptide binding. Notably, this list of methods and descriptions of structure-based approaches is not exhaustive. For a more comprehensive review of this topic, please refer to [92].
As was mentioned above, some neoantigens that have a high binding affinity to MHC will not be effectively recognized by the TCR [100][101], which makes them unable to trigger T cell-mediated immune response. This fact allows us to suggest the existence of some peptide features that determine their recognition by the TCR independently of MHC binding. In recently published works, it was reported that immunogenic peptides are enriched in hydrophobic and aromatic amino acids at positions interacting with the TCR [102][103]. Other parameters that are believed to influence TCR binding are amino acid charge and bulkiness, WT and mutant sequence divergence and sequence entropy [2][104]. Currently, available tools attempt to solve these challenges by considering these features in the context of the peptide sequence [2][103][104][105]. However, it is evident that the impact of properties such as amino acid charge and size and the composition of hydrophobic residues should be taken into account in the conformation of the peptide bound to the MHC. In this connection, structure-based predictions could be one of the possible ways to determine the impact of physicochemical features of peptides on their immunogenic potential [91][106][107]. In [107], the authors developed a flexible backbone docking protocol called TCRFlexDock utilizing RosettaDock and ZRANK and benchmarked it using 20 structures of TCR/pMHC (17 for MHC class I and 3 for MHC class II) complexes, for which resolved structures of unbound components are available. Testing revealed that protein–protein docking algorithms are able to produce accurate structural models of TCR/pMHC based on unbound component structures [107]. In [106], the authors used a force-field approach utilizing refined versions of FoldX and Rosetta force fields to perform prediction of related targets of the TCR. TCR:p:MHCII complex-based benchmark containing epitope and non-epitope containing pMHC complexes was developed, and immunogenicity was estimated by calculating interaction energies between the TCR and each of the p:MHCII complexes. It was found that the predictive power of this approach depends on the ability to predict protein-MHC complex binding and model the structure of the TCR:p:MHC complex [106]. Riley et al. [91] developed a procedure for accurate and rapid modeling of the structure of nonameric peptides bound with a common class I MHC type HLA-A2 and applied it for analyzing a dataset containing thousands of immunogenic, non-immunogenic and non-HLA2-A2 binding peptides. After that, they trained a neural network (NN) on structural features that affect TCR and peptide binding energies. It was shown that structurally-parameterized NN outperformed other methods that do not include explicit structural or energetic properties in the assessment of CD8+ T cell response of HLA-A2 bound nonameric peptides [91]. Thus, a combination of MHC-binding prediction based on NGS-data with a structure-based approach could significantly improve the accuracy of immunogenic peptide selection that is of special importance in the context of peptide-based cancer vaccine development.
References
- Hundal, J.; Kiwala, S.; McMichael, J.; Miller, C.A.; Xia, H.; Wollam, A.T.; Liu, C.J.; Zhao, S.; Feng, Y.Y.; Graubert, A.P.; et al. pVACtools: A computational toolkit to identify and visualize cancer neoantigens. Cancer Immunol. Res. 2020, 8, 409.
- Kim, S.; Kim, H.S.; Kim, E.; Lee, M.G.; Shin, E.C.; Paik, S.; Kim, S. Neopepsee: Accurate genome-level prediction of neoantigens by harnessing sequence and amino acid immunogenicity information. Ann. Oncol. Off. J. Eur. Soc. Med. Oncol. 2018, 29, 1030–1036.
- Rubinsteyn, A.; Hodes, I.; Kodysh, J.; Hammerbacher, J. Vaxrank: A computational tool for designing personalized cancer vaccines. bioRxiv 2017, 142919.
- Rubinsteyn, A.; Kodysh, J.; Hodes, I.; Mondet, S.; Aksoy, B.A.; Finnigan, J.P.; Bhardwaj, N.; Hammerbacher, J. Computational pipeline for the PGV-001 neoantigen vaccine trial. Front. Immunol. 2018, 8.
- Schenck, R.O.; Lakatos, E.; Gatenbee, C.; Graham, T.A.; Anderson, A.R.A. Neopredpipe: High-throughput neoantigen prediction and recognition potential pipeline. BMC Bioinf. 2019, 20, 264.
- Richters, M.M.; Xia, H.; Campbell, K.M.; Gillanders, W.E.; Griffith, O.L.; Griffith, M. Best practices for bioinformatic characterization of neoantigens for clinical utility. Genome Med. 2019, 11, 56.
- Turajlic, S.; Litchfield, K.; Xu, H.; Rosenthal, R.; McGranahan, N.; Reading, J.L.; Wong, Y.N.S.; Rowan, A.; Kanu, N.; Al Bakir, M.; et al. Insertion-and-deletion-derived tumour-specific neoantigens and the immunogenic phenotype: A pan-cancer analysis. Lancet Oncol. 2017, 18, 1009–1021.
- Yang, W.; Lee, K.W.; Srivastava, R.M.; Kuo, F.; Krishna, C.; Chowell, D.; Makarov, V.; Hoen, D.; Dalin, M.G.; Wexler, L.; et al. Immunogenic neoantigens derived from gene fusions stimulate T cell responses. Nat. Med. 2019, 25, 767–775.
- David, J.K.; Maden, S.K.; Weeder, B.R.; Thompson, R.F.; Nellore, A. Putatively cancer-specific exon–exon junctions are shared across patients and present in developmental and other non-cancer cells. NAR Cancer 2020, 2.
- Smart, A.C.; Margolis, C.A.; Pimentel, H.; He, M.X.; Miao, D.; Adeegbe, D.; Fugmann, T.; Wong, K.K.; Van Allen, E.M. Intron retention is a source of neoepitopes in cancer. Nat. Biotechnol. 2018, 36, 1056–1058.
- Park, J.; Chung, Y.J. Identification of neoantigens derived from alternative splicing and RNA modification. Genom. Inform. 2019, 17, e23.
- Shen, L.; Zhang, J.; Lee, H.; Batista, M.T.; Johnston, S.A. RNA Transcription and splicing errors as a source of cancer frameshift neoantigens for vaccines. Sci. Rep. 2019, 9, 14184.
- Laumont, C.M.; Vincent, K.; Hesnard, L.; Audemard, E.; Bonneil, E.; Laverdure, J.P.; Gendron, P.; Courcelles, M.; Hardy, M.P.; Cote, C.; et al. Noncoding regions are the main source of targetable tumor-specific antigens. Sci. Trans. Med. 2018, 10.
- Khodadoust, M.S.; Olsson, N.; Wagar, L.E.; Haabeth, O.A.; Chen, B.; Swaminathan, K.; Rawson, K.; Liu, C.L.; Steiner, D.; Lund, P.; et al. Antigen presentation profiling reveals recognition of lymphoma immunoglobulin neoantigens. Nature 2017, 543, 723–727.
- Khodadoust, M.S.; Olsson, N.; Chen, B.; Sworder, B.; Shree, T.; Liu, C.L.; Zhang, L.; Czerwinski, D.K.; Davis, M.M.; Levy, R.; et al. B-cell lymphomas present immunoglobulin neoantigens. Blood 2019, 133, 878–881.
- Walboomers, J.M.; Jacobs, M.V.; Manos, M.M.; Bosch, F.X.; Kummer, J.A.; Shah, K.V.; Snijders, P.J.; Peto, J.; Meijer, C.J.; Muñoz, N. Human papillomavirus is a necessary cause of invasive cervical cancer worldwide. J. Pathol. 1999, 189, 12–19.
- Gillison, M.L.; Koch, W.M.; Capone, R.B.; Spafford, M.; Westra, W.H.; Wu, L.; Zahurak, M.L.; Daniel, R.W.; Viglione, M.; Symer, D.E.; et al. Evidence for a causal association between human papillomavirus and a subset of head and neck cancers. JNCI J. Natl. Cancer Inst. 2000, 92, 709–720.
- Liepe, J.; Marino, F.; Sidney, J.; Jeko, A.; Bunting, D.E.; Sette, A.; Kloetzel, P.M.; Stumpf, M.P.; Heck, A.J.; Mishto, M. A large fraction of HLA class I ligands are proteasome-generated spliced peptides. Science (NY) 2016, 354, 354–358.
- Kumai, T.; Ishibashi, K.; Oikawa, K.; Matsuda, Y.; Aoki, N.; Kimura, S.; Hayashi, S.; Kitada, M.; Harabuchi, Y.; Celis, E.; et al. Induction of tumor-reactive T helper responses by a posttranslational modified epitope from tumor protein p53. Cancer Immunol. Immunother. Cii 2014, 63, 469–478.
- Wood, M.A.; Nguyen, A.; Struck, A.J.; Ellrott, K.; Nellore, A.; Thompson, R.F. Neoepiscope improves neoepitope prediction with multivariant phasing. Bioinformatics (Oxf. Engl.) 2019, 36, 713–720.
- Zhao, Q.; Laverdure, J.P.; Lanoix, J.; Durette, C.; Coté, C.; Bonneil, E.; Laumont, C.M.; Gendron, P.; Vincent, K.; Courcelles, M.; et al. Proteogenomics uncovers a vast repertoire of shared tumor-specific antigens in ovarian cancer. Cancer Immunol. Res. 2020.
- Chen, Z.; Yuan, Y.; Chen, X.; Chen, J.; Lin, S.; Li, X.; Du, H. Systematic comparison of somatic variant calling performance among different sequencing depth and mutation frequency. Sci. Rep. 2020, 10, 3501.
- Warden, C.D.; Adamson, A.W.; Neuhausen, S.L.; Wu, X. Detailed comparison of two popular variant calling packages for exome and targeted exon studies. PeerJ 2014, 2, e600.
- Alioto, T.S.; Buchhalter, I.; Derdak, S.; Hutter, B.; Eldridge, M.D.; Hovig, E.; Heisler, L.E.; Beck, T.A.; Simpson, J.T.; Tonon, L.; et al. A comprehensive assessment of somatic mutation detection in cancer using whole-genome sequencing. Nat. Commun. 2015, 6, 10001.
- Callari, M.; Sammut, S.J.; De Mattos-Arruda, L.; Bruna, A.; Rueda, O.M.; Chin, S.F.; Caldas, C. Intersect-then-combine approach: Improving the performance of somatic variant calling in whole exome sequencing data using multiple aligners and callers. Genome Med. 2017, 9, 35.
- Saunders, C.T.; Wong, W.S.; Swamy, S.; Becq, J.; Murray, L.J.; Cheetham, R.K. Strelka: Accurate somatic small-variant calling from sequenced tumor-normal sample pairs. Bioinformatics (Oxf. Engl.) 2012, 28, 1811–1817.
- Shiraishi, Y.; Sato, Y.; Chiba, K.; Okuno, Y.; Nagata, Y.; Yoshida, K.; Shiba, N.; Hayashi, Y.; Kume, H.; Homma, Y.; et al. An empirical Bayesian framework for somatic mutation detection from cancer genome sequencing data. Nucleic Acids Res. 2013, 41, e89.
- Ye, K.; Schulz, M.H.; Long, Q.; Apweiler, R.; Ning, Z. Pindel: A pattern growth approach to detect break points of large deletions and medium sized insertions from paired-end short reads. Bioinformatics (Oxf. Engl.) 2009, 25, 2865–2871.
- Zhang, J.; White, N.M.; Schmidt, H.K.; Fulton, R.S.; Tomlinson, C.; Warren, W.C.; Wilson, R.K.; Maher, C.A. Integrate: Gene fusion discovery using whole genome and transcriptome data. Genome Res. 2016, 26, 108–118.
- Zhang, J.; Mardis, E.R.; Maher, C.A. INTEGRATE-neo: A pipeline for personalized gene fusion neoantigen discovery. Bioinformatics (Oxf. Engl.) 2017, 33, 555–557.
- Haas, B.J.; Dobin, A.; Stransky, N.; Li, B.; Yang, X.; Tickle, T.; Bankapur, A.; Ganote, C.; Doak, T.G.; Pochet, N.; et al. STAR-Fusion: Fast and accurate fusion transcript detection from RNA-Seq. bioRxiv 2017.
- Sijts, E.J.A.M.; Kloetzel, P.M. The role of the proteasome in the generation of MHC class I ligands and immune responses. Cell. Mol. Life Sci. CMLS 2011, 68, 1491–1502.
- Rock, K.L.; Reits, E.; Neefjes, J. Present yourself! By MHC Class I and MHC Class II molecules. Trends Immunol. 2016, 37, 724–737.
- Peters, B.; Bulik, S.; Tampe, R.; Van Endert, P.M.; Holzhütter, H.G. Identifying MHC class I epitopes by predicting the TAP transport efficiency of epitope precursors. J. Immunol. (Baltimore, Md. 1950) 2003, 171, 1741–1749.
- Bhasin, M.; Raghava, G.P.S. Analysis and prediction of affinity of TAP binding peptides using cascade SVM. Protein Sci. 2004, 13, 596–607.
- Tenzer, S.; Peters, B.; Bulik, S.; Schoor, O.; Lemmel, C.; Schatz, M.M.; Kloetzel, P.M.; Rammensee, H.G.; Schild, H.; Holzhütter, H.G. Modeling the MHC class I pathway by combining predictions of proteasomal cleavage, TAP transport and MHC class I binding. Cell. Mol. Life Sci. CMLS 2005, 62, 1025–1037.
- Bhasin, M.; Lata, S.; Raghava, G.P. TAPPred prediction of TAP-binding peptides in antigens. Methods Mol. Biol. 2007, 409, 381–386.
- Nielsen, M.; Lundegaard, C.; Lund, O.; Keşmir, C. The role of the proteasome in generating cytotoxic T-cell epitopes: Insights obtained from improved predictions of proteasomal cleavage. Immunogenetics 2005, 57, 33–41.
- Hoze, E.; Tsaban, L.; Maman, Y.; Louzoun, Y. Predictor for the effect of amino acid composition on CD4 + T cell epitopes preprocessing. J. Immunol. Methods 2013, 391, 163–173.
- Paul, S.; Karosiene, E.; Dhanda, S.K.; Jurtz, V.; Edwards, L.; Nielsen, M.; Sette, A.; Peters, B. Determination of a predictive cleavage motif for eluted major histocompatibility complex class II ligands. Front. Immunol. 2018, 9, 1795.
- Romero, J.M.; Jiménez, P.; Cabrera, T.; Cózar, J.M.; Pedrinaci, S.; Tallada, M.; Garrido, F.; Ruiz-Cabello, F. Coordinated downregulation of the antigen presentation machinery and HLA class I/beta2-microglobulin complex is responsible for HLA-ABC loss in bladder cancer. Int. J. Cancer 2005, 113, 605–610.
- Leone, P.; Shin, E.C.; Perosa, F.; Vacca, A.; Dammacco, F.; Racanelli, V. MHC class I antigen processing and presenting machinery: Organization, function, and defects in tumor cells. J. Natl. Cancer Inst. 2013, 105, 1172–1187.
- Yewdell, J.W.; Reits, E.; Neefjes, J. Making sense of mass destruction: Quantitating MHC class I antigen presentation. Nat. Rev. Immunol. 2003, 3, 952–961.
- Melista, E.; Rigo, K.; Pasztor, A.; Christiansen, M.; Bertinetto, F.E.; Meintjes, P.; Hague, T. Towards a new gold standard—NGS corrections to sanger SBT genotyping results. Hum. Immunol. 2015, 76, 148.
- Choo, S.Y. The HLA system: Genetics, immunology, clinical testing, and clinical implications. Yonsei Med. J. 2007, 48, 11–23.
- Bauer, D.C.; Zadoorian, A.; Wilson, L.O.W.; Thorne, N.P. Evaluation of computational programs to predict HLA genotypes from genomic sequencing data. Brief. Bioinf. 2018, 19, 179–187.
- Kiyotani, K.; Mai, T.H.; Nakamura, Y. Comparison of exome-based HLA class I genotyping tools: Identification of platform-specific genotyping errors. J. Hum. Genet. 2017, 62, 397–405.
- Paulson, K.G.; Voillet, V.; McAfee, M.S.; Hunter, D.S.; Wagener, F.D.; Perdicchio, M.; Valente, W.J.; Koelle, S.J.; Church, C.D.; Vandeven, N.; et al. Acquired cancer resistance to combination immunotherapy from transcriptional loss of class I HLA. Nat. Commun. 2018, 9, 3868.
- Paulson, K.G.; Tegeder, A.; Willmes, C.; Iyer, J.G.; Afanasiev, O.K.; Schrama, D.; Koba, S.; Thibodeau, R.; Nagase, K.; Simonson, W.T.; et al. Downregulation of MHC-I expression is prevalent but reversible in Merkel cell carcinoma. Cancer Immunol. Res. 2014, 2, 1071–1079.
- McGranahan, N.; Rosenthal, R.; Hiley, C.T.; Rowan, A.J.; Watkins, T.B.K.; Wilson, G.A.; Birkbak, N.J.; Veeriah, S.; Van Loo, P.; Herrero, J.; et al. Allele-Specific HLA Loss and immune escape in lung cancer evolution. Cell 2017, 171, 1259–1271.e1211.
- Reynisson, B.; Alvarez, B.; Paul, S.; Peters, B.; Nielsen, M. NetMHCpan-4.1 and NetMHCIIpan-4.0: Improved predictions of MHC antigen presentation by concurrent motif deconvolution and integration of MS MHC eluted ligand data. Nucleic Acids Res. 2020, 48, W449–W454.
- Jurtz, V.; Paul, S.; Andreatta, M.; Marcatili, P.; Peters, B.; Nielsen, M. NetMHCpan-4.0: Improved peptide-MHC class I interaction predictions integrating eluted ligand and peptide binding affinity data. J. Immunol. 2017, 199, 3360–3368.
- O’Donnell, T.J.; Rubinsteyn, A.; Laserson, U. MHCflurry 2.0: Improved pan-allele prediction of MHC Class I-presented peptides by incorporating antigen processing. Cell Syst. 2020, 11, P42–P48.e7.
- O’Donnell, T.J.; Rubinsteyn, A.; Bonsack, M.; Riemer, A.B.; Laserson, U.; Hammerbacher, J. MHCflurry: Open-source class I MHC binding affinity prediction. Cell Syst. 2018, 7, 129–132.e124.
- Reynisson, B.; Barra, C.; Kaabinejadian, S.; Hildebrand, W.H.; Peters, B.; Nielsen, M. Improved prediction of MHC II antigen presentation through integration and motif deconvolution of mass spectrometry MHC eluted ligand data. J. Proteome Res. 2020, 19, 2304–2315.
- Bonsack, M.; Hoppe, S.; Winter, J.; Tichy, D.; Zeller, C.; Küpper, M.; Schitter, E.C.; Blatnik, R.; Riemer, A.B. Performance evaluation of MHC class-I binding prediction tools based on an experimentally validated MHC-peptide binding dataset. Cancer Immunol. Res. 2019.
- Zhao, W.; Sher, X. Systematically benchmarking peptide-MHC binding predictors: From synthetic to naturally processed epitopes. PLoS Comput. Biol. 2018, 14, e1006457.
- Paul, S.; Croft, N.P.; Purcell, A.W.; Tscharke, D.C.; Sette, A.; Nielsen, M.; Peters, B. Benchmarking predictions of MHC class I restricted T cell epitopes. bioRxiv 2019, 694539.
- Trolle, T.; Metushi, I.G.; Greenbaum, J.A.; Kim, Y.; Sidney, J.; Lund, O.; Sette, A.; Peters, B.; Nielsen, M. Automated benchmarking of peptide-MHC class I binding predictions. Bioinformaticsatics (Oxf. Engl.) 2015, 31, 2174–2181.
- Paul, S.; Croft, N.P.; Purcell, A.W.; Tscharke, D.C.; Sette, A.; Nielsen, M.; Peters, B. Benchmarking predictions of MHC class I restricted T cell epitopes in a comprehensively studied model system. PLoS Comput. Biol. 2020, 16, e1007757.
- Croft, N.P.; Smith, S.A.; Pickering, J.; Sidney, J.; Peters, B.; Faridi, P.; Witney, M.J.; Sebastian, P.; Flesch, I.E.A.; Heading, S.L.; et al. Most viral peptides displayed by class I MHC on infected cells are immunogenic. Proc. Natl. Acad. Sci. USA 2019, 116, 3112–3117.
- Sette, A.; Vitiello, A.; Reherman, B.; Fowler, P.; Nayersina, R.; Kast, W.M.; Melief, C.J.; Oseroff, C.; Yuan, L.; Ruppert, J.; et al. The relationship between class I binding affinity and immunogenicity of potential cytotoxic T cell epitopes. J. Immunol. (Baltimore, Md. 1950) 1994, 153, 5586–5592.
- Bjerregaard, A.M.; Nielsen, M.; Jurtz, V.; Barra, C.M.; Hadrup, S.R.; Szallasi, Z.; Eklund, A.C. An Analysis of natural T cell responses to predicted tumor neoepitopes. Front. Immunol. 2017, 8, 1566.
- Castle, J.C.; Kreiter, S.; Diekmann, J.; Löwer, M.; van de Roemer, N.; de Graaf, J.; Selmi, A.; Diken, M.; Boegel, S.; Paret, C.; et al. Exploiting the mutanome for tumor vaccination. Cancer Res. 2012, 72, 1081.
- Bekri, S.; Uduman, M.; Gruenstein, D.; Mei, A.H.C.; Tung, K.; Rodney-Sandy, R.; Bogen, B.; Buell, J.; Stein, R.; Doherty, K.; et al. Neoantigen synthetic peptide vaccine for multiple myeloma elicits T cell immunity in a pre-clinical model. Blood 2017, 130, 1868.
- Kreiter, S.; Vormehr, M.; van de Roemer, N.; Diken, M.; Löwer, M.; Diekmann, J.; Boegel, S.; Schrörs, B.; Vascotto, F.; Castle, J.C.; et al. Mutant MHC class II epitopes drive therapeutic immune responses to cancer. Nature 2015, 520, 692–696.
- Borg, N.A.; Ely, L.K.; Beddoe, T.; Macdonald, W.A.; Reid, H.H.; Clements, C.S.; Purcell, A.W.; Kjer-Nielsen, L.; Miles, J.J.; Burrows, S.R.; et al. The CDR3 regions of an immunodominant T cell receptor dictate the ‘energetic landscape’ of peptide-MHC recognition. Nat. Immunol. 2005, 6, 171–180.
- Gras, S.; Saulquin, X.; Reiser, J.B.; Debeaupuis, E.; Echasserieau, K.; Kissenpfennig, A.; Legoux, F.; Chouquet, A.; Le Gorrec, M.; Machillot, P.; et al. Structural bases for the affinity-driven selection of a public TCR against a dominant human cytomegalovirus epitope. J. Immunol. (Baltimore, Md. 1950) 2009, 183, 430–437.
- Chen, G.; Yang, X.; Ko, A.; Sun, X.; Gao, M.; Zhang, Y.; Shi, A.; Mariuzza, R.A.; Weng, N.P. Sequence and structural analyses reveal distinct and highly diverse human cd8(+) tcr repertoires to immunodominant viral antigens. Cell Rep. 2017, 19, 569–583.
- Nivarthi, U.K.; Gras, S.; Kjer-Nielsen, L.; Berry, R.; Lucet, I.S.; Miles, J.J.; Tracy, S.L.; Purcell, A.W.; Bowden, D.S.; Hellard, M.; et al. An extensive antigenic footprint underpins immunodominant TCR adaptability against a hypervariable viral determinant. J. Immunol. (Baltimore, Md. 1950) 2014, 193, 5402–5413.
- Dash, P.; Fiore-Gartland, A.J.; Hertz, T.; Wang, G.C.; Sharma, S.; Souquette, A.; Crawford, J.C.; Clemens, E.B.; Nguyen, T.H.O.; Kedzierska, K.; et al. Quantifiable predictive features define epitope-specific T cell receptor repertoires. Nature 2017, 547, 89–93.
- Gielis, S.; Moris, P.; Neuter, N.D.; Bittremieux, W.; Ogunjimi, B.; Laukens, K.; Meysman, P. TCRex: A webtool for the prediction of T-cell receptor sequence epitope specificity. bioRxiv 2018, 373472.
- Jurtz, V.I.; Jessen, L.E.; Bentzen, A.K.; Jespersen, M.C.; Mahajan, S.; Vita, R.; Jensen, K.K.; Marcatili, P.; Hadrup, S.R.; Peters, B.; et al. NetTCR: Sequence-based prediction of TCR binding to peptide-MHC complexes using convolutional neural networks. bioRxiv 2018, 433706.
- Ogishi, M.; Yotsuyanagi, H. Quantitative prediction of the Landscape of T cell epitope immunogenicity in sequence space. Front. Immunol. 2019, 10.
- Springer, I.; Besser, H.; Tickotsky-Moskovitz, N.; Dvorkin, S.; Louzoun, Y. Prediction of specific TCR-peptide binding from large dictionaries of TCR-peptide pairs. bioRxiv 2020, 650861.
- Bi, J.; Zheng, Y.; Yan, F.; Hou, S.; Li, C. Prediction of epitope-associated TCR by using network topological similarity based on deepwalk. IEEE Access 2019, 7, 151273–151281.
- Zhang, Y.; Lin, Z.; Wan, Y.; Cai, H.; Deng, L.; Li, R. The Immunogenicity and anti-tumor efficacy of a rationally designed neoantigen vaccine for B16F10 mouse melanoma. Front. Immunol. 2019, 10, 2472.
- Ni, Q.; Zhang, F.; Liu, Y.; Wang, Z.; Yu, G.; Liang, B.; Niu, G.; Su, T.; Zhu, G.; Lu, G.; et al. A bi-adjuvant nanovaccine that potentiates immunogenicity of neoantigen for combination immunotherapy of colorectal cancer. Sci. Adv. 2020, 6, eaaw6071.
- Chen, R.; Fulton, K.M.; Twine, S.M.; Li, J. Identification of MHC Peptides Using Mass Spectrometry For Neoantigen Discovery And Cancer Vaccine Development. Mass. Spectrum. Rev. 2019.
- Zhang, X.; Qi, Y.; Zhang, Q.; Liu, W. Application of mass spectrometry-based MHC immunopeptidome profiling in neoantigen identification for tumor immunotherapy. Biomed. Pharmacother. 2019, 120, 109542.
- Creech, A.L.; Ting, Y.S.; Goulding, S.P.; Sauld, J.F.K.; Barthelme, D.; Rooney, M.S.; Addona, T.A.; Abelin, J.G. The role of mass spectrometry and proteogenomics in the advancement of HLA epitope prediction. Proteomics 2018, 18, e1700259.
- Storkus, W.J.; Zeh, H.J., 3rd; Salter, R.D.; Lotze, M.T. Identification of T-cell epitopes: Rapid isolation of class I-presented peptides from viable cells by mild acid elution. J. Immunother. Emphas. Tumor Immunol. Off. J. Soc. Biol. Ther. 1993, 14, 94–103.
- Kote, S.; Pirog, A.; Bedran, G.; Alfaro, J.; Dapic, I. Mass Spectrometry-based identification of MHC-associated peptides. Cancers 2020, 12, 535.
- Vigneron, N.; Stroobant, V.; Ferrari, V.; Abi Habib, J.; Van den Eynde, B.J. Production of spliced peptides by the proteasome. Mol. Immunol. 2019, 113, 93–102.
- Liepe, J.; Sidney, J.; Lorenz, F.K.M.; Sette, A.; Mishto, M. Mapping the MHC class I–spliced immunopeptidome of cancer cells. Cancer Immunol. Res. 2019, 7, 62.
- Mylonas, R.; Beer, I.; Iseli, C.; Chong, C.; Pak, H.S.; Gfeller, D.; Coukos, G.; Xenarios, I.; Müller, M.; Bassani-Sternberg, M. Estimating the contribution of proteasomal spliced peptides to the HLA-I ligandome. Mol. Cell Proteom. 2018, 17, 2347.
- Engelhard, V.H.; Altrich-Vanlith, M.; Ostankovitch, M.; Zarling, A.L. Post-translational modifications of naturally processed MHC-binding epitopes. Curr. Opin. Immunol. 2006, 18, 92–97.
- Solleder, M.; Guillaume, P.; Racle, J.; Michaux, J.; Pak, H.; Müller, M.; Coukos, G.; Bassani-Sternberg, M.; Gfeller, D. Mass spectrometry based immunopeptidomics leads to robust predictions of phosphorylated HLA class I ligands. bioRxiv 2019, 836189.
- Bulik-Sullivan, B.; Busby, J.; Palmer, C.D.; Davis, M.J.; Murphy, T.; Clark, A.; Busby, M.; Duke, F.; Yang, A.; Young, L.; et al. Deep learning using tumor HLA peptide mass spectrometry datasets improves neoantigen identification. Nat. Biotechnol. 2019, 37, 55–63.
- Li, Y.; Wang, G.; Tan, X.; Ouyang, J.; Zhang, M.; Song, X.; Liu, Q.; Leng, Q.; Chen, L.; Xie, L. ProGeo-neo: A customized proteogenomic workflow for neoantigen prediction and selection. BMC Med. Genom. 2020, 13, 52.
- Riley, T.P.; Keller, G.L.J.; Smith, A.R.; Davancaze, L.M.; Arbuiso, A.G.; Devlin, J.R.; Baker, B.M. Structure based prediction of neoantigen immunogenicity. Front. Immunol. 2019, 10.
- Antunes, D.A.; Abella, J.R.; Devaurs, D.; Rigo, M.M.; Kavraki, L.E. Structure-based methods for binding mode and binding affinity prediction for peptide-MHC complexes. Curr. Top. Med. Chem. 2018, 18, 2239–2255.
- Mohammed, F.; Stones, D.H.; Zarling, A.L.; Willcox, C.R.; Shabanowitz, J.; Cummings, K.L.; Hunt, D.F.; Cobbold, M.; Engelhard, V.H.; Willcox, B.E. The antigenic identity of human class I MHC phosphopeptides is critically dependent upon phosphorylation status. Oncotarget 2017, 8, 54160–54172.
- Durrant, L.G.; Metheringham, R.L.; Brentville, V.A. Autophagy, citrullination and cancer. Autophagy 2016, 12, 1055–1056.
- Galli-Stampino, L.; Meinjohanns, E.; Frische, K.; Meldal, M.; Jensen, T.; Werdelin, O.; Mouritsen, S. T-cell recognition of tumor-associated carbohydrates: The nature of the glycan moiety plays a decisive role in determining glycopeptide immunogenicity. Cancer Res. 1997, 57, 3214–3222.
- Schueler-Furman, O.; Altuvia, Y.; Sette, A.; Margalit, H. Structure-based prediction of binding peptides to MHC class I molecules: Application to a broad range of MHC alleles. Protein Sci. 2000, 9, 1838–1846.
- Bui, H.H.; Schiewe, A.J.; von Grafenstein, H.; Haworth, I.S. Structural prediction of peptides binding to MHC class I molecules. Proteins 2006, 63, 43–52.
- Yanover, C.; Bradley, P. Large-scale characterization of peptide-MHC binding landscapes with structural simulations. Proc. Natl. Acad. Sci. USA 2011, 108, 6981.
- Mukherjee, S.; Bhattacharyya, C.; Chandra, N. HLaffy: Estimating peptide affinities for Class-1 HLA molecules by learning position-specific pair potentials. Bioinformatics (Oxf. Engl.) 2016, 32, 2297–2305.
- Ochoa-Garay, J.; McKinney, D.M.; Kochounian, H.H.; McMillan, M. The ability of peptides to induce cytotoxic T cells in vitro does not strongly correlate with their affinity for the H-2Ld molecule: Implications for vaccine design and immunotherapy. Mol. Immunol. 1997, 34, 273–281.
- Feltkamp, M.C.; Vierboom, M.P.; Kast, W.M.; Melief, C.J. Efficient MHC class I-peptide binding is required but does not ensure MHC class I-restricted immunogenicity. Mol. Immunol. 1994, 31, 1391–1401.
- Calis, J.J.; Maybeno, M.; Greenbaum, J.A.; Weiskopf, D.; De Silva, A.D.; Sette, A.; Kesmir, C.; Peters, B. Properties of MHC class I presented peptides that enhance immunogenicity. PLoS Comput. Biol. 2013, 9, e1003266.
- Chowell, D.; Krishna, S.; Becker, P.D.; Cocita, C.; Shu, J.; Tan, X.; Greenberg, P.D.; Klavinskis, L.S.; Blattman, J.N.; Anderson, K.S. TCR contact residue hydrophobicity is a hallmark of immunogenic CD8+ T cell epitopes. Proc. Nat. Acad. Sci. USA 2015, 112, E1754–E1762.
- Tung, C.W.; Ziehm, M.; Kämper, A.; Kohlbacher, O.; Ho, S.Y. POPISK: T-cell reactivity prediction using support vector machines and string kernels. BMC Bioinf. 2011, 12, 446.
- Trolle, T.; Nielsen, M. NetTepi: An integrated method for the prediction of T cell epitopes. Immunogenetics 2014, 66, 449–456.
- Lanzarotti, E.; Marcatili, P.; Nielsen, M. Identification of the cognate peptide-MHC target of T cell receptors using molecular modeling and force field scoring. Mol. Immunol. 2018, 94, 91–97.
- Pierce, B.G.; Weng, Z. A flexible docking approach for prediction of T cell receptor-peptide-MHC complexes. Protein Sci. 2013, 22, 35–46.

