 +1 credit
+1 credit
Video Upload Options
Authors describe how by coupling emerging in silico and experimental tools it is possible to create novel peptide libraries with potential antimicrobial activity. This is in response to the growing public health concern pose by multiresistant microbial strains that take millions of lives annually on a global scale. The in silico tools include emerging artificial intelligence algorithms that allow searching for novel sequences in extremely large databases. Once identified, the required membrane activity can be estimated by looking at the interactions with model lipid bilayers via molecular dynamics (MD) simulations. Experimentally, the sequences can be expressed on the surface of yeasts by the surface display technology and subsequently screened in a high-throughput manner aided by microfluidic systems capable of separating out the most active peptides by precisely monitoring changes in optical properties in-line and real-time.
1. Introduction
Antimicrobial resistance (AMR), both inherent and acquired, has become an issue of increasing concern in recent years. AMR negatively impacts population health and healthcare systems costs and gross domestic product (GDP) [1]. Inherent resistance is a natural attribute that protects the organism from antimicrobials (AM), such as the Gram-negative bacteria’s outer membrane. Contrarily, acquired resistance is caused by genetic mutations that enable the microorganism to resist antimicrobials through different underlying mechanisms. Within those mechanisms, some of the most important include drug inactivation by enzymes, cell wall modifications, alteration of AM targets’ binding sites, efflux pumps that expel the AM bypassing the targets, and modification of metabolic pathways [2]. AMR is the consequence of misuse and overuse of antibiotics, self-medication, self-interrupted treatments, exposure to nosocomial infections in hospitals, genetic plasticity, and sheer dogged adaptability of the microorganisms themselves [2][3]. To complicate the situation even further, the pharmaceutical industry has virtually stopped developing new antibiotics, mainly due to economic and regulatory obstacles. By 2015, 15 out of 18 of the largest pharmaceutical companies had abandoned the antibiotic field [4]. This lack of new molecules to treat infections has been related to a new pre-antibiotic era, in which infections and minor injuries which have been treatable for decades may once again kill millions [5]. Recent studies have estimated that by 2050, more than ten million deaths per year are attributed to resistant pathogens, with a higher percentage of them occurring in developing countries [6].
Specifically, for bacteria, by 2016, the World Health Organization (WHO) reported that the global incidence of infection cases approached 490,000, which can be attributed to these resistant pathogens [5]. In 2019, just in the USA, more than 2.8 million antibiotic-resistant infections were reported by the CDC (Center for Disease Control and Prevention), which resulted in more than 35,000 deaths [7]. These statistics clearly show the exponential increase in resistant bacteria. The ESKAPE group comprises six nosocomial multidrug-resistant microorganisms (Enterococcus faecium, Staphylococcus aureus, Klebsiella pneumoniae, Acinetobacter baumannii, Pseudomonas aeruginosa, and Enterobacter spp.) and it is listed by the WHO as a priority to acquired new antibiotics. Within this group, the most problematic ones are perhaps the carbapenem (last resort family of antibiotics) resistant A. baumannii, P. aeruginosa, K. pneumoniae, and Enterobacter spp. and for that reason, they are listed with critical priority. Additionally, concerning are the vancomycin-resistant E. faecium and methicillin and vancomycin-resistant S. aureus, which are listed with high priority. ESKAPE pathogens are responsible for most nosocomial infections and represent the vast majority of isolates whose resistance presents serious therapeutic dilemmas to physicians such as experimental treatment selection, comorbidities treatment, especially cancer, and isolation procedures [3][8]. Compared to non-ESKAPE pathogens, the ESKAPE group has shown a higher mortality rate and higher costs due to the need for more comprehensive and sophisticated treatments [9].
Furthermore, there are reports of resistance incidences against some of the more newly discovered/designed antibiotics, and the outlook appears not to improve in the coming years. Therefore, an imperative is to find alternative treatments, especially for the ESKAPE pathogens [3]. Moreover, there is a concern because these infections are no longer confined to hospitals. Over recent years, rising resistant infections in the community have been detected, which can put more people at risk, in addition to making the spread more challenging to identify and contain [10].
Virus resistance is a less concerning issue. However, some important or common viruses, such as influenza, hepatitis C, herpes, and human immunodeficiency virus (HIV), exhibit AM resistance. HIV has shown inherent resistance to some antiretrovirals (ARV) via some proteases and reverse transcriptases [11]. Some countries have recently reported AM levels at or above 15% among patients starting ARV treatment and 40% among patients after re-starting treatment. This shows that HIV has also acquired resistance, which has led to substantial economic implications, given that second and third-line treatments are three times and 18 times more expensive, respectively [5]. Influenza virus has a high mutation rate; therefore, it easily achieves resistance to most commonly used antivirals such as adamantanes and the neuraminidase inhibitors (NAIs). Furthermore, in 2018 resistance to favipiravir, a broad-spectrum antiviral, was reported in vitro, suggesting that a possible unreported resistance mechanism could exist in the worldwide population [12].
An alternative treatment for all the resistant microorganisms described above has emerged in the last decade: the antimicrobial peptides (AMPs). AMPs are short peptides with a broad spectrum of antimicrobial activities that are part of living organisms’ defense mechanisms against microbial pathogens. Since AMPs have diverse chemical features and cellular targets, they are promising AM agents with an expected lower rate of acquired resistance by microorganisms [13]. The action of most antimicrobial peptides relies on the interaction between the positive charges in the peptide’s residues and the negatively charged membrane components. The structural and physicochemical properties of antimicrobial peptides and their capacity to adopt an amphipathic conformation upon membrane binding influence this interaction [13]. This conformation results from a balance between positively charged and hydrophobic amino acid residues [14]. The insertion of antimicrobial peptides into the membrane’s hydrophobic core depends on the microbe membrane [13].
Regarding bacteria, the interaction with AMPs varies between Gram-negative and Gram-positive microorganisms. Cationic AMPs have shown to cross the outer membrane of Gram-negative bacteria by a charge-exchange mechanism of competition with membrane-bound Ca2+ and Mg2+. Upon interaction, the peptides bind to lipopolysaccharides, most likely promoted by the binding to outer membrane proteins, thereby reaching the cell membrane [15]. In contrast, given the cell wall’s porosity of Gram-positive bacteria, many AMPs seem to pass relatively easily. Once on the cell surface, single-cell studies have shown the accumulation of AMPs to be restricted to foci associated with cell division, cell wall remodeling, or secretion, thereby interfering with these vital processes or causing cell lysis [16]. Cationic AMPs amphipathic conformation allows increased interaction with the negatively charged surfaces or direct insertion into the bacterial membranes. Additionally, the higher potential inside the negative transmembrane in bacteria further enhances the strength of electrostatic attraction. The AMP–membrane interaction has been typically associated with barrel-stave, carpet, or toroidal-pore models [14].
Studies have shown that a single peptide can act through several mechanisms mediated by the topology, aggregation, and lipid interactions of AMPs with cellular membranes. These, in turn, rely on the peptide structure, the peptide/lipid ratio, and the properties of the lipid membrane [17]. Additionally, depending on AMPs concentration, the cellular membrane can rather expand, which results in pores that allow the transport of the peptide into the microorganism or generate local or massive ruptures of the membrane. Local perturbances induced by the peptides are due to interactions with proteins, nucleic acids, and cellular organelles, which by itself constitutes a potential cell-killing mechanism. The ability of individual AMPs to interact with multiple targets or multiple peptides to interact with a single target may limit the development of bacterial resistance [14].
Nowadays, peptides discovery is facilitated through library screening via both rational and non-rational approaches. There are three major methods for rational design: template-based design, physicochemical, and de novo methods, aiming to create novel peptides and/or improve existing ones. The template-based design aims to add selectivity and/or increase a known peptide sequence activity by including an amino acid or changing its position. This generally results in a reduction in the peptide sizes. With this approach, it is possible to identify novel AMP sequences even from inactive peptides. The physicochemical design also generates analogs with different physicochemical properties from known sequences. Finally, the de novo method creates new peptides based on amino acid patterns or frequencies [18].
The de novo method creates new peptides based on amino acid patterns or frequencies [18]. This approach is based on identifying sequence patterns, crucial residue positions, and amino acid frequencies from known AMPs. This information is then used to develop prediction methods and linguistic models to identify novel AMPs [19]. Generally, de novo AMP design involves favoring an amphipathic structure such that the peptide sequences exhibit both hydrophobic and hydrophilic regions [20]. Furthermore, de novo design can also be completed aided by machine learning methods such as variational autoencoders (VAEs) and generative adversarial networks (GANs). In the case of VAEs, the input data serves as the basis to create a continuous latent space that can be used to further interpolate between objects. As a result, by interpolating between two known AMPs it is possible to generate novel chemical structures that represent a smooth transition between both peptides. Dean and colleagues used this approach to obtain novel AMPs that where successfully validated experimentally [21]. In the case of GANs, the distribution of the input peptides is followed to generate a set of new ones through a two machine learning networks: the generator, that generates the new peptide sequences and the discriminator, whose task is to try to discriminate between real and fake peptides. In this way, at the end of the training the generated example peptides should not be discriminated as false and exhibit similar properties to the real ones [22].
In contrast, non-rational approaches rely on the microbial surface display technology for obtaining various well-established random peptide sequences for further screening according to the available methods within the framework of the peptide-based drug discovery process [23]. Combinatorial chemistry has been used to create libraries of peptides/proteins and discover new recombinant therapeutics [24]. Combinatorial library methods can be generated by vastly diverse chemical libraries, including phage display, yeast display, bacteria display, mRNA display, and more [25]. This technique’s primary strength is its capability to generate the enormously diverse exogenous peptides or proteins displayed on the cell’s surface using standard yet rapid molecular biology methods instead of using genetically engineered protein or peptide variants individually [26]. Guralp and colleagues proposed a five-step light-directed in situ parallel oligonucleotide synthesis with a cellular expression and screening system. The first step involves the AMP library design, where designed peptides and reverse-translated to oligonucleotides. In the second step, the library is synthesized by parallel synthesis technology which allows large numbers of oligonucleotides to be produced on a single array. In the third step, emulsion PCR is employed to amplify each of the oligonucleotides followed by their cloning and expression to find the bioactive peptide sequences. Finally, microorganism strains showing AMPs activities are chosen and their plasmids extracted for DNA sequencing and subsequent identification of the AMP candidates in silico [27]. Peptides have profoundly impacted the modern pharmaceutical industry’s development and have contributed significantly to biological and chemical science [24].
Furthermore, phage display technology, a combinatorial screening approach, provides a molecular diversity tool [24] for creating libraries of random peptides and proteins to identify ligands for receptors, identify enzyme blockers, studying protein/DNA–protein interactions, screening cDNA expression, epitope mapping of antibodies, engineering human antibodies, optimizing antibody specificities, identifying peptides that home to specific organs or tissues, generating immunogens for vaccine design, and use in affinity chromatography [26]. Phage display has several advantages over traditional random screening methods used in drug discovery, such as simplicity, cost-effectiveness, and speed [26].
The cell-surface display allows peptides to be displayed on microbial cells’ surface by fusing them with the anchoring motifs, usually cell-surface proteins or their fragments. The fusion can be accomplished by N-terminal fusion, C-terminal fusion, or sandwich fusion. The characteristics of carrier protein, passenger protein, and the host cell and fusion method might impact the efficiency of surface display of proteins [28].
Given the potential of AMPs to address the worldwide concern on resistant organisms, our research group proposes both rational and non-rational frameworks for a more efficient and faster method to find antimicrobial peptides. Our approaches rely on bacteria/yeasts surface display and low-cost microfluidics for screening such that experimentation costs are reduced without compromising throughput (Figure 1). Regarding rational design, the framework consists of a four-step process that aims to minimize time and resources, taking only the most promising peptides to experimental evaluation (Figure 1). The workflow begins with two computational phases: the first one comprises deep learning techniques to find sequences with potential antimicrobial activity. In the second one, the candidates are subjected to interaction with a cell membrane in silico via molecular dynamics (MD). This approach allows us to identify whether a candidate has the membrane-disruption capabilities required to be an AMP. Subsequently, the sequence is passed to the experimental phase where (I) the host with which the analysis will be carried out is modified by following the current molecular biology methods for surface display, and (II) a microfluidic system is used to corroborate the antimicrobial activity. Alternatively, for non-rational design, the framework consists of a three-step process: (I) Cell surface display, followed by (II) microfluidics analysis, and (III) DNA sequencing.
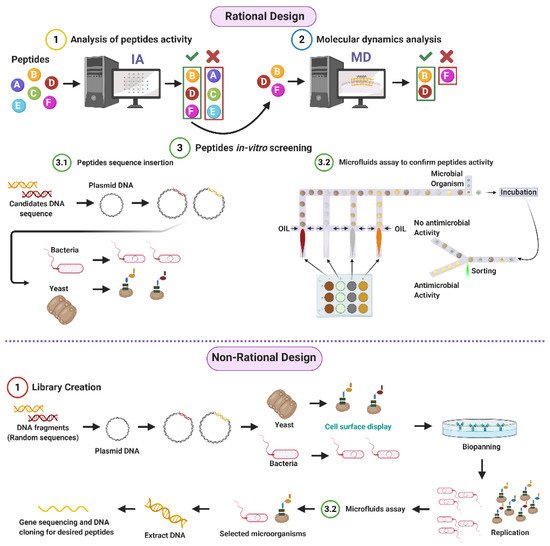
Figure 1. Antimicrobial Peptides (AMPs) discovery framework. Rational design steps: (I) Deep learning techniques identify sequences with potential antimicrobial activity, (II) membrane-disruption capabilities of selected sequences are analyzed via molecular dynamics (MD), (III) the host cell is modified, and sequences are inserted, finally (IV) antimicrobial activity is corroborated by a microfluidic system. Non-rational design steps: (I) Random sequences are expressed on host cells through cell surface display, (II) modified microorganisms are analyzed by a microfluidics system to obtain AMPs candidates, and (III) DNA is extracted, sequenced, and cloned (Created with BioRender).
The first stage’s deep learning algorithm is based on recurrent neural networks (RNNs) composed of several layers, enabling learning data representations with multiple abstraction levels. The algorithm was inspired by natural language processing (NLP) techniques considering their suitability for problems based on the sequence’s involved elements. In this way, the generated representations can be easily interconverted into simpler ones. The reliability of these architectures has been previously demonstrated in property prediction and generation of molecules with certain features of interest [29][30][31][32]. The initial layers of the RNNs are capable of learning local information, while the deeper layers are focused more on learning global and abstract information [33]. For example, the initial layers will learn features representing functional groups or amino acids present in the peptides. In contrast, the deeper layers will learn features related to the amino acids’ sequence and the peptide’s global structure, which will enable predictions about their biological activity. The deeper layers take the initial layers’ as input information and combine them through mathematical operations to achieve that level of abstraction. Finally, for the learning process to be possible, a backpropagation algorithm is implemented to minimize an error function established at the beginning of the training process by adjusting each layer’s internal parameters iteratively [33].
Regarding the second stage, peptides–membrane interaction analysis computational simulations provide a powerful tool to understand different molecules’ properties through their interaction at a molecular and nanoscale [34]. These simulations provide missing information on the mechanistic details at the molecular scale of such interactions. Therefore, this approach closes a knowledge gap concerning the macroscopic information collected experimentally [35]. Moreover, it provides additional insights into controversial or counterintuitive results obtained at the macroscopic scale [36]. To achieve an understanding of the system at the atomic level, diverse techniques have been used, where Monte Carlo (MC) [37][38] and molecular dynamics (MD) rank high among the preferred choices [39][40]. These methodologies emerged in the late 1950s when Alder and Wainright published the first description of these tools, which were used to analyze the phase transition for hard-sphere systems [41]. Since then, they have evolved, becoming more accessible and powerful and reaching out to various research areas, including chemistry, materials science, biology, geology, and physics [42][43]. The main goals are to understand the interactions among several molecules involved in a particular situation and guide new experimental strategies toward a desired state by the insights provided by the simulations [44].
MC simulations have attracted significant attention for a deeper understanding of interactions due to their versatility. They allow us to calculate multiple solutions with multiple unknowns, with a simple program structure and its relative ease of implementation [45]. MC simulations are essentially based on non-deterministic models that assign random numbers to trajectories associated with the atoms’ displacements [46]. The Metropolis Monte Carlo (MMC) has become very popular over the years because its use is not restricted only to states of equilibrium but can be extended to calculating dynamic properties [47]. This approach searches for an equilibrium state of the system within probable states generated by a Boltzmann distribution [46]. A second technique with high importance corresponds to the molecular dynamic simulations, which allows determining the equilibrium and transport properties by finding the atoms’ displacement through a numerical solution of Newton’s equations of motion [34]. Some of the most used algorithms in MD correspond to the Verlet, velocity Verlet, and Leapfrog algorithms, which satisfy the symplectic condition [48].
Currently available software packages for MD simulations popular include AMBER [49], GROMACS [50], CHARMM [51], NAMD [52], LAMMPS [53], and DL-POLY [54]. The first four software packages are principally developed for biochemical macromolecules such as proteins, lipids, and nucleic acids. Simultaneously, LAMMPS is focused on materials modeling, and DL-POLY is a general-purpose simulation package [54][55]. The difference between them mainly lies in their performance, capacity, data processing, and adaptability to new hardware. For instance, coupling to GPUs of exceedingly high performance should be easily achievable to shorten simulation times significantly [56].
MD simulations have demonstrated exceedingly high performance in finding information at the atomic level in silico that would be very difficult to obtain experimentally [57]. In the context of our work, this is the case of peptide–lipid bilayer interactions. Therefore, the collected information is valuable to investigate different aspects of such interactions, including the mechanism of action and the toxicity of peptides with antimicrobial and other membrane activities [58]. Moreover, it is possible to conduct experiments in different lipid membrane models, such as bacterial, mammalian, and even carcinogenic [59]. Additionally, diseases involving dependence on the composition of the bilayer, such as cancer, Alzheimer’s, and cardiovascular diseases, can be explored mechanistically in silico to guide the experimental development of novel therapeutic approaches [60][61][62]. An example of a classical representation of a peptide–lipid bilayer system in MD is given in Figure 2.
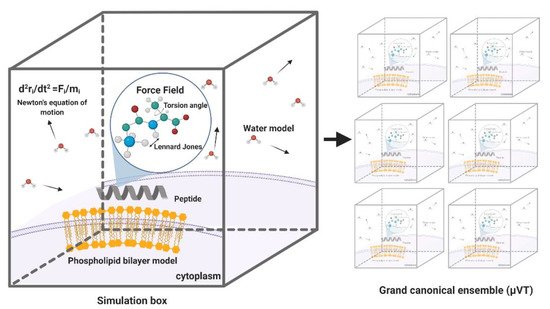
Figure 2. Representation of a traditional membrane-protein system used in molecular dynamics. The interaction among the components is modeled through force fields that account for variations in key parameters and impose restrictions on the accessible states (Created with BioRender).
The last stage of our proposed framework is dedicated to screening potential candidates experimentally via microfluidics platforms. This microsystem family has been comprehensively explored to screen different bioactive compounds, including DNA, proteins, enzymes, receptors, and peptides [63]. The development of platforms for single cells screening, to produce biofuels and drug screening resistance assays [64][65]; biomarkers, involved in the reliable prediction of diseases [66]; screening of bacteria with high production of lactic acid such as Bacillus coagulans [67] and library screening for enzyme engineering applications [68][69], are proof of the versatility of this mechanism, showing promising results in the field of biotechnology. In all cases, this approach has been considered advantageous, mainly due to the ability to perform thousands of reactions at the nanoliter to femtoliter scale, replacing robotic automation using small volume samples, reducing unit costs of experimentation, and increasing throughput [70][71][72]. Additionally, microfluidics offers a dynamic integration with different components, allowing the interaction between several variables within a single platform, providing the tools to increase the assays’ precision, accurate determination, and control of experimental conditions. Finally, the ability to handle features in the range of a single cell proportion results in scaled down readouts and a single cell resolution sensitivity [65][70][71][73]. Remarkably, in peptides, microfluidics has reduced reagents utilization and sample consumption, provided shorter times, and fully automatized the process [74]. The implemented microfluidics screening techniques for the case of antimicrobial peptides include three main strategies, namely, droplet-based, membrane-based, and combinatorial microarrays, which are explained in more detail below.
2. Antimicrobial Peptides
Antimicrobial peptides (AMPs) represent essential components of the higher organisms’ innate immunity; however, they are produced by all lifeforms [75]. AMPs have been isolated from microorganisms, fungi, insects, and other invertebrates, plants, amphibians, birds, fish, and mammals, including humans. These peptides are produced either by ribosomal translation of mRNA or by nonribosomal peptide synthesis, mainly identified in bacteria [75]. AMPs are short sequences (12 to 100 amino acids) that generally exhibit broad-spectrum activity and cationic behavior with a net charge ranging from +2 to +9. Additionally, they are usually amphipathic and, in most cases, present hydrophobicity levels greater than 30% [76]. Lysine, arginine, tryptophan, and cysteine residues are highly conserved throughout their structure. Lysine and arginine have been thought responsible for enabling electrostatic interactions between the peptide and negatively charged membranes.
Additionally, given tryptophans’ unique sidechain containing an indole ring that holds hydrogen-bonding potential, they show strong membrane-disruptive activities by interacting with a membrane’s interface capable of anchoring the peptide to the surface of the bilayer. Regarding cysteine, the disulfide bonds formed are strongly hydrophobic and play an essential role in the peptides’ overall structure and increasing stability towards proteolytic degradation [77]. Given the wide range of antimicrobial activity and varied action mechanisms, AMPs are currently under study as alternative biomolecules to treat infections in scenarios involving resistant microorganisms. Several antimicrobial peptides have been reported in various databases such as The Collection of Anti-Microbial Peptides CAMPR3 (8164 entries) [78], Database of Antimicrobial Activity and Structure of Peptides DBAASP v3.0 (16180 entries) [79] and The Data Repository of Antimicrobial Peptides DRAMP v2.0 (19899 entries) [80]. These peptides can be categorized by their origin, either synthetic or natural, by taxonomy and by activity. According to the DRAMP database, activity classification is divided into four principal classes, antibacterial (7856), antiviral (2015), antifungal (3371) and antiparasitic (148), but also into Anti-Gram+ (2568), Anti-Gram- (2397), anticancer (293), antitumor (156), insecticidal (246) and antiprotozoal (17).
2.1. Antibacterial
AMPs with antibacterial activity are the most studied. Antibacterial peptides can be classified into non-ribosomal synthetic peptides and natural or synthetic ribosomal peptides [81]. The first group is mainly produced by bacteria, while the last is produced by all animals and bacteria [82]. Virtually all antibacterial peptides have less than 100 amino acid residues, mainly in the range of three to 50 [83]. The antibacterial peptides structure has four styles, including α helices, β-sheet, extended and looped shapes. The β sheet and the α helix are more abundant in nature [84]. Most of them are cationic with hydrophilic and hydrophobic domains, allowing them to target bacterial cell membranes and cause the lipid bilayer structure’s breakdown. Furthermore, AMPs can kill bacteria by inhibiting some important cell pathways, such as DNA replication and protein synthesis [85].
Many researchers believe that the ability of AMPS to bind to bacterial membranes plays a vital role in their development [86][87]. Some mechanisms for attaching AMP to bacterial membranes include the cane, the toroidal pore wormhole, the carpet pattern, and detergent [76]. The main obstacle in using antibacterial peptides is their ability to lyse eukaryotic cells, especially red blood cells. For their application, they must have low hemolytic activity and high antimicrobial activity [88].
2.2. Antivirals
Antiviral peptides are biochemically characterized by being cationic and amphipathic, with net positive charges to effectively work as antimicrobials. Different reports reveal that hydrophobicity seems to be a fundamental property to assure significant activity against enveloped viruses [89]. Antiviral peptides are classified according to their mechanism of action [90]. This includes blocking viral receptors, inhibiting adsorption by antimicrobial binding peptides to viral proteins, interaction with co-receptors such as CXCR4, inhibition of cell fusion by interfering with the protein’s ATPase activity, inhibition of gene expression, inhibition of peptide elongation, and activation of immunomodulatory pathways [18][91][92].
2.3. Antifungal
Most antifungal peptides (AFPs) exhibit rapid and potent membrane activity and show a low likelihood of inducing de novo resistance given their wide range of inhibitory mechanisms. As for the other AMPs, AFPs are produced by all living organisms. When generated by unicellular organisms, they are small with a structure containing non-protein amino acids and a fatty acyl moiety. Simultaneously, the AFPs produced by multicellular organisms are more extensive, with the majority having either linear α-helical or cystine-stabilized defensin-like structures. AFPs can be divided structurally into linear peptides, β-sheet peptides, peptides with a mixture of α-helices and β-sheets, and peptides rich in amino acids specific moieties such as modified cyclic peptides, depsipeptides, and lipopeptides [93]. Alternatively, AFPs can also be classified by their action mechanism as membrane-disrupting lytic peptides, which are usually amphipathic and abundant in nature. Cell wall synthesis or bio-synthesis obstructive AFPs are safe and effective for immune-compromised patients [94]. AFPs have also been incorporated into food formulations for preservation purposes [95].
2.4. Antiparasitic
Antiparasitic peptides (APPs) are by far the least studied ones. For this reason, there is no recollection of their structural similarities with the other families of AMPs. However, many peptides such as defensins, scorpines, decoralins, drosomycins, cecropins, and Buforin II have been reported as antiparasitic [96][97][98]. For a review on APPs, we encourage the reader to consult [98]. In general, the APP’s action mechanism is associated with selective parasite’s membrane disruption, which usually takes place within the host cell where the parasite is often hidden. Once APPs bind to the host’s membrane, the peptide can transfer to the parasite membrane and exert a lytic activity. Such transferring ability is attributed to the parasite infection’s permeability pathways into the host cells [96].
References
- Naylor, N.R.; Atun, R.; Zhu, N.; Kulasabanathan, K.; Silva, S.; Chatterjee, A.; Knight, G.M.; Robotham, J.V. Estimating the burden of antimicrobial resistance: A systematic literature review. Antimicrob. Resist. Infect. Control 2018, 7.
- Stokowski, L.A. Antimicrobial Resistance: A Primer. Available online: (accessed on 3 November 2020).
- Mulani, M.S.; Kamble, E.E.; Kumkar, S.N.; Tawre, M.S.; Pardesi, K.R. Emerging Strategies to Combat ESKAPE Pathogens in the Era of Antimicrobial Resistance: A Review. Front. Microbiol. 2019, 10, 539.
- Ventola, C.L. The Antibiotic Resistance Crisis: Part 1: Causes and Threats. Pharm. Ther. 2015, 40, 277–283.
- World Health Organization (WHO). Antimicrobial Resistance. Available online: (accessed on 3 November 2020).
- Sakeena, M.H.F.; Bennett, A.A.; McLachlan, A.J. Enhancing pharmacists’ role in developing countries to overcome the challenge of antimicrobial resistance: A narrative review. Antimicrob. Resist. Infect. Control 2018, 7.
- Center for Disease Control and Prevention (CDC). Antibiotic-Resistant Germs: New Threats. Available online: (accessed on 3 November 2020).
- El-Mahallawy, H.A.; Hassan, S.S.; El-Wakil, M.; Moneer, M.M. Bacteremia due to ESKAPE pathogens: An emerging problem in cancer patients. J. Egypt. Natl. Cancer Inst. 2016, 28, 157–162.
- Marturano, J.E.; Lowery, T.J. ESKAPE Pathogens in Bloodstream Infections Are Associated with Higher Cost and Mortality but Can Be Predicted Using Diagnoses Upon Admission. Open Forum Infect. Dis. 2019, 6.
- Pendleton, J.N.; Gorman, S.P.; Gilmore, B.F. Clinical relevance of the ESKAPE pathogens. Expert Rev. Anti Infect. Ther. 2013, 11, 297–308.
- Tang, M.W.; Shafer, R.W. HIV-1 Antiretroviral Resistance. Drugs 2012, 72, e1–e25.
- Goldhill, D.H.; Te Velthuis, A.J.; Fletcher, R.A.; Langat, P.; Zambon, M.; Lackenby, A.; Barclay, W.S. The mechanism of resistance to favipiravir in influenza. Proc. Natl. Acad. Sci. USA 2018, 115, 11613–11618.
- Lázár, V.; Martins, A.; Spohn, R.; Daruka, L.; Grézal, G.; Fekete, G.; Számel, M.; Jangir, P.K.; Kintses, B.; Csörgő, B.; et al. Antibiotic-resistant bacteria show widespread collateral sensitivity to antimicrobial peptides. Nat. Microbiol. 2018, 3, 718–731.
- Bechinger, B.; Gorr, S.-U. Antimicrobial Peptides: Mechanisms of Action and Resistance. J. Dent. Res. 2016, 96, 254–260.
- Anunthawan, T.; De La Fuente-Núñez, C.; Hancock, R.E.; Klaynongsruang, S. Cationic amphipathic peptides KT2 and RT2 are taken up into bacterial cells and kill planktonic and biofilm bacteria. Biochim. Biophys. Acta Biomembr. 2015, 1848, 1352–1358.
- Malanovic, N.; Lohner, K. Gram-positive bacterial cell envelopes: The impact on the activity of antimicrobial peptides. Biochim. Biophys. Acta Biomembr. 2016, 1858, 936–946.
- Bechinger, B. The SMART model: Soft Membranes Adapt and Respond also Transiently, in the presence of antimicrobial peptides. J. Pept. Sci. 2014, 21, 346–355.
- Boas, L.C.P.V.; Campos, M.L.; Berlanda, R.L.A.; de Carvalho Neves, N.; Franco, O.L. Antiviral peptides as promising therapeutic drugs. Cell. Mol. Life Sci. 2019, 76, 3525–3542.
- Waghu, F.H.; Joseph, S.; Ghawali, S.; Martis, E.A.; Madan, T.; Venkatesh, K.V.; Idicula-Thomas, S. Designing antibacterial peptides with enhanced killing kinetics. Front. Microbiol. 2018, 9, 325.
- Torres, M.D.; Sothiselvam, S.; Lu, T.K.; de la Fuente-Nunez, C. Peptide design principles for antimicrobial applications. J. Mol. Biol. 2019, 431, 3547–3567.
- Dean, S.N.; Walper, S.A. Variational Autoencoder for Generation of Antimicrobial Peptides. ACS Omega 2020, 5, 20746–20754.
- Lin, E.; Lin, C.H.; Lane, H.Y. Relevant Applications of Generative Adversarial Networks in Drug Design and Discovery: Molecular De Novo Design Dimensionality Reduction, and De Novo Peptide and Protein Design. Molecules 2020, 25, 3250.
- Kalafatovic, D.; Mauša, G.; Todorovski, T.; Giralt, E. Algorithm-supported, mass and sequence diversity-oriented random peptide library design. J. Cheminform. 2019, 11, 25.
- Henninot, A.; Collins, J.C.; Nuss, J.M. The current state of peptide drug discovery: Back to the future? J. Med. Chem. 2018, 61, 1382–1414.
- Liu, R.; Li, X.; Lam, K.S. Combinatorial chemistry in drug discovery. Curr. Opin. Chem. Biol. 2017, 38, 117–126.
- Hamzeh-Mivehroud, M.; Alizadeh, A.A.; Morris, M.B.; Church, W.B.; Dastmalchi, S. Phage display as a technology delivering on the promise of peptide drug discovery. Drug Discov. Today 2013, 18, 1144–1157.
- Guralp, S.A.; Murgha, Y.E.; Rouillard, J.M.; Gulari, E. From design to screening: A new antimicrobial peptide discovery pipeline. PLoS ONE 2013, 8, e59305.
- Lee, S.Y.; Choi, J.H.; Xu, Z. Microbial cell-surface display. Trends Biotechnol. 2003, 21, 45–52.
- Lane, N.; Kahanda, I. DeepACPpred: A Novel Hybrid CNN-RNN Architecture for Predicting Anti-Cancer Peptides. In Advances in Intelligent Systems and Computing; Springer International Publishing: Cham, Switzerland, 2020; pp. 60–69.
- Müller, A.T.; Hiss, J.A.; Schneider, G. Recurrent Neural Network Model for Constructive Peptide Design. J. Chem. Inf. Model. 2018, 58, 472–479.
- Goh, G.B.; Siegel, C.; Vishnu, A.; Hodas, N. Using Rule-Based Labels for Weak Supervised Learning. In Proceedings of the 24th ACM SIGKDD International Conference on Knowledge Discovery & Data Mining, London, UK, 19–23 August 2018; Association for Computing Machinery: New York, NY, USA, 2018.
- Tiwary, S.; Levy, R.; Gutenbrunner, P.; Soto, F.S.; Palaniappan, K.K.; Deming, L.; Berndl, M.; Brant, A.; Cimermancic, P.; Cox, J. High-quality MS/MS spectrum prediction for data-dependent and data-independent acquisition data analysis. Nat. Methods 2019, 16, 519–525.
- Goodfellow, I.; Bengio, Y.; Courville, A.; Bengio, Y. Deep Learning; MIT Press Cambridge: Cambridge, MA, USA, 2016; Volume 1.
- Wahnström, G. Molecular Dynamics Lecture Notes; Chalmers University of Technology: Gothenburg, Sweden, 2018.
- Feig, M.; Nawrocki, G.; Yu, I.; Wang, P.-H.; Sugita, Y. Challenges and opportunities in connecting simulations with experiments via molecular dynamics of cellular environments. J. Phys. Conf. Ser. 2018, 1036.
- Allen, M.P. Introduction to Molecular Dynamics Simulation. In Computational Soft Matter: From Synthetic Polymers to Proteins; John von Neumann Institute for Computing (NIC): Jülich, Germany, 2004.
- Rathore, N.; Pablo, J.J. de Monte Carlo simulation of proteins through a random walk in energy space. J. Chem. Phys. 2002, 116, 7225–7230.
- Gofman, Y.; Haliloglu, T.; Ben-Tal, N. Monte-Carlo Simulations of Peptide-Membrane Interactions: Web-Server. Biophys. J. 2010, 98, 487a.
- Karplus, M.; McCammon, J.A. Molecular dynamics simulations of biomolecules. Nat. Struct. Biol. 2002, 9, 646–652.
- Reif, M.; Zacharias, M. Computer Modelling and Molecular Dynamics Simulation of Biomolecules. In Biomolecular and Bioanalytical Techniques; John Wiley & Sons Ltd.: New York, NY, USA, 2019; pp. 501–535.
- Alder, B.J.; Wainwright, T.E. Phase Transition for a Hard Sphere System. J. Chem. Phys. 1957, 27, 1208–1209.
- Hollingsworth, S.A.; Dror, R.O. Molecular Dynamics Simulation for All. Neuron 2018, 99, 1129–1143.
- Hernández, E.R.; Zetina, L.M.M.; Vega, G.T.; Rocha, M.G.; Ochoa, L.F.R.; Fernandez, R.L. Molecular Dynamics: From basic techniques to applications (A Molecular Dynamics Primer). In AIP Conference Proceedings; AIP: College Park, MD, USA, 2008.
- Aliaga, L.C.R.; Lima, L.V.P.C.; Domingues, G.M.B.; Bastos, I.N.; Evangelakis, G.A. Experimental and molecular dynamics simulation study on the glass formation of Cu-Zr-Al alloys. Mater. Res. Express 2019, 6.
- Chen, J. The Development and Comparison of Molecular Dynamics Simulation and Monte Carlo Simulation. IOP Conf. Ser. Earth Environ. Sci. 2018, 128.
- Neyts, E.C.; Bogaerts, A. Combining molecular dynamics with Monte Carlo simulations: Implementations and applications. Theor. Chem. Acc. Belg. 2012, 132.
- Kikuchi, K.; Yoshida, M.; Maekawa, T.; Watanabe, H. Metropolis Monte Carlo method as a numerical technique to solve the FokkerPlanck equation. Chem. Phys. Lett. 1991, 185, 335–338.
- Cuendet, M.A.; van Gunsteren, W.F. On the calculation of velocity-dependent properties in molecular dynamics simulations using the leapfrog integration algorithm. J. Chem. Phys. 2007, 127.
- Case, D.A.; Cheatham, T.E.; Darden, T.; Gohlke, H.; Luo, R.; Merz, K.M.; Onufriev, A.; Simmerling, C.; Wang, B.; Woods, R.J. The Amber biomolecular simulation programs. J. Comput. Chem. 2005, 26, 1668–1688.
- Spoel, D.V.D.; Lindahl, E.; Hess, B.; Groenhof, G.; Mark, A.E.; Berendsen, H.J.C. GROMACS: Fast flexible, and free. J. Comput. Chem. 2005, 26, 1701–1718.
- Brooks, B.R.; Brooks, C.L.; Mackerell, A.D.; Nilsson, L.; Petrella, R.J.; Roux, B.; Won, Y.; Archontis, G.; Bartels, C.; Boresch, S.; et al. CHARMM: The biomolecular simulation program. J. Comput. Chem. 2009, 30, 1545–1614.
- Phillips, J.C.; Braun, R.; Wang, W.; Gumbart, J.; Tajkhorshid, E.; Villa, E.; Chipot, C.; Skeel, R.D.; Kalé, L.; Schulten, K. Scalable molecular dynamics with NAMD. J. Comput. Chem. 2005, 26, 1781–1802.
- Plimpton, S. Fast Parallel Algorithms for Short-Range Molecular Dynamics. J. Comput. Phys. 1995, 117, 1–19.
- Smith, W.; Yong, C.W.; Rodger, P.M. DL_POLY: Application to molecular simulation. Mol. Simul. 2002, 28, 385–471.
- FrantzDale, B.; Plimpton, S.J.; Shephard, M.S. Software components for parallel multiscale simulation: An example with LAMMPS. Eng. Comput. 2009, 26, 205–211.
- Hernández-Rodríguez, M.; Rosales-Hernández, M.C.; Mendieta-Wejebe, J.E.; Martínez-Archundia, M.; Basurto, J.C. Current Tools and Methods in Molecular Dynamics (MD) Simulations for Drug Design. Curr. Med. Chem. 2016, 23, 3909–3924.
- Martinez-Seara, H.; Róg, T. Molecular Dynamics Simulations of Lipid Bilayers: Simple Recipe of How to Do It. In Methods in Molecular Biology; Humana Press: Totowa, NJ, USA, 2012; pp. 407–429.
- Langham, A.; Kaznessis, Y.N. Molecular Simulations of Antimicrobial Peptides. In Methods in Molecular Biology; Humana Press: Totowa, NJ, USA, 2009; pp. 267–285.
- Shahane, G.; Ding, W.; Palaiokostas, M.; Orsi, M. Physical properties of model biological lipid bilayers: Insights from all-atom molecular dynamics simulations. J. Mol. Model. 2019, 25.
- Bharadwaj, P.; Solomon, T.; Malajczuk, C.J.; Mancera, R.L.; Howard, M.; Arrigan, D.W.M.; Newsholme, P.; Martins, R.N. Role of the cell membrane interface in modulating production and uptake of Alzheimers beta amyloid protein. Biochim. Biophys. Acta Biomembr. 2018, 1860, 1639–1651.
- Szlasa, W.; Zendran, I.; Zalesińska, A.; Tarek, M.; Kulbacka, J. Lipid composition of the cancer cell membrane. J. Bioenerg. Biomembr. 2020, 52, 321–342.
- Revin, V.V.; Gromova, N.V.; Revina, E.S.; Martynova, M.I.; Seikina, A.I.; Revina, N.V.; Imarova, O.G.; Solomadin, I.N.; Tychkov, A.Y.; Zhelev, N. Role of Membrane Lipids in the Regulation of Erythrocytic Oxygen-Transport Function in Cardiovascular Diseases. BioMed Res. Int. 2016, 2016, 3429604.
- Fu, Y.; Luo, J.; Qin, J.; Yang, M. Screening techniques for the identification of bioactive compounds in natural products. J. Pharm. Biomed. Anal. 2019, 168, 189–200.
- Kim, H.S.; Hsu, S.-C.; Han, S.-I.; Thapa, H.R.; Guzman, A.R.; Browne, D.R.; Tatli, M.; Devarenne, T.P.; Stern, D.B.; Han, A. High-throughput droplet microfluidics screening platform for selecting fast-growing and high lipid-producing microalgae from a mutant library. Plant Direct 2017, 1, e00011.
- Barata, D.; van Blitterswijk, C.; Habibovic, P. High-throughput screening approaches and combinatorial development of biomaterials using microfluidics. Acta Biomater. 2016, 34, 1–20.
- Kaushik, A.M.; Hsieh, K.; Wang, T.-H. Droplet microfluidics for high-sensitivity and high-throughput detection and screening of disease biomarkers. Wiley Interdiscip. Rev. Nanomed. Nanobiotechnol. 2018, 10.
- Zhu, X.-D.; Shi, X.; Wang, S.-W.; Chu, J.; Zhu, W.-H.; Ye, B.-C.; Zuo, P.; Wang, Y.-H. High-throughput screening of high lactic acid-producing Bacillus coagulans by droplet microfluidic based flow cytometry with fluorescence activated cell sorting. RSC Adv. 2019, 9, 4507–4513.
- Longwell, C.K.; Labanieh, L.; Cochran, J.R. High-throughput screening technologies for enzyme engineering. Curr. Opin. Biotechnol. 2017, 48, 196–202.
- Prodanović, R.; Ung, W.L.; Đurđić, K.I.; Fischer, R.; Weitz, D.A.; Ostafe, R. A high-throughput screening system based on droplet microfluidics for glucose oxidase gene libraries. Molecules 2020, 25, 2418.
- Mashaghi, S.; Abbaspourrad, A.; Weitz, D.A.; van Oijen, A.M. Droplet microfluidics: A tool for biology chemistry and nanotechnology. TrAC Trends Anal. Chem. 2016, 82, 118–125.
- Li, X.; Yang, X.; Liu, L.; Zhou, P.; Zhou, J.; Shi, X.; Wang, Y. A microarray platform designed for high-throughput screening the reaction conditions for the synthesis of micro/nanosized biomedical materials. Bioact. Mater. 2020, 5, 286–296.
- Holland-Moritz, D.A.; Wismer, M.K.; Mann, B.F.; Farasat, I.; Devine, P.; Guetschow, E.D.; Mangion, I.; Welch, C.J.; Moore, J.C.; Sun, S.; et al. Mass Activated Droplet Sorting (MADS) Enables High-Throughput Screening of Enzymatic Reactions at Nanoliter Scale. Angew. Chem. Int. Ed. 2020, 59, 4470–4477.
- Lim, J.W.; Shin, K.S.; Moon, J.; Lee, S.K.; Kim, T. A Microfluidic Platform for High-Throughput Screening of Small Mutant Libraries. Anal. Chem. 2016, 88, 5234–5242.
- Che, Y.-J.; Wu, H.-W.; Hung, L.-Y.; Liu, C.-A.; Chang, H.-Y.; Wang, K.; Lee, G.-B. An integrated microfluidic system for screening of phage-displayed peptides specific to colon cancer cells and colon cancer stem cells. Biomicrofluidics 2015, 9.
- Mahlapuu, M.; Håkansson, J.; Ringstad, L.; Björn, C. Antimicrobial Peptides: An Emerging Category of Therapeutic Agents. Front. Cell. Infect. Microbiol. 2016, 6.
- Kumar, P.; Kizhakkedathu, J.; Straus, S. Antimicrobial Peptides: Diversity Mechanism of Action and Strategies to Improve the Activity and Biocompatibility In Vivo. Biomolecules 2018, 8, 4.
- Mojsoska, B.; Jenssen, H. Peptides and Peptidomimetics for Antimicrobial Drug Design. Pharmaceuticals 2015, 8, 366–415.
- Waghu, F.H.; Barai, R.S.; Gurung, P.; Idicula-Thomas, S. CAMPR3: A database on sequences, structures and signatures of antimicrobial peptides: Table 1. Nucleic Acids Res. 2015, 44, D1094–D1097.
- Pirtskhalava, M.; Amstrong, A.A.; Grigolava, M.; Chubinidze, M.; Alimbarashvili, E.; Vishnepolsky, B.; Gabrielian, A.; Rosenthal, A.; Hurt, D.E.; Tartakovsky, M. DBAASP v3: Database of antimicrobial/cytotoxic activity and structure of peptides as a resource for development of new therapeutics. Nucleic Acids Res. 2020.
- Kang, X.; Dong, F.; Shi, C.; Liu, S.; Sun, J.; Chen, J.; Li, H.; Xu, H.; Lao, X.; Zheng, H. DRAMP 2.0, an updated data repository of antimicrobial peptides. Sci. Data 2019, 6.
- Zheng, Z.; Tharmalingam, N.; Liu, Q.; Jayamani, E.; Kim, W.; Fuchs, B.B.; Zhang, R.; Vilcinskas, A.; Mylonakis, E. Synergistic efficacy of Aedes aegypti antimicrobial peptide cecropin A2 and tetracycline against Pseudomonas aeruginosa. Antimicrob. Agents Chemother. 2017, 61.
- Martin, G.E.; Boudreau, R.M.; Couch, C.; Becker, K.A.; Edwards, M.J.; Caldwell, C.C.; Gulbins, E.; Seitz, A. Sphingosine’s role in epithelial host defense: A natural antimicrobial and novel therapeutic. Biochimie 2017, 141, 91–96.
- Sedaghati, M.; Ezzatpanah, H.; Boojar, M.M.A.; Ebrahimi, M.T.; Kobarfard, F. Isolation and identification of some antibacterial peptides in the plasmin-digest of ββββ-casein. LWT Food Sci. Technol. 2016, 68, 217–225.
- Harmouche, N.; Aisenbrey, C.; Porcelli, F.; Xia, Y.; Nelson, S.E.D.; Chen, X.; Raya, J.; Vermeer, L.; Aparicio, C.; Veglia, G.; et al. Solution and solid-state nuclear magnetic resonance structural investigations of the antimicrobial designer peptide GL13K in membranes. Biochemistry 2017, 56, 4269–4278.
- Speck-Planche, A.; Kleandrova, V.V.; Ruso, J.M.; DS Cordeiro, M.N. First multitarget chemo-Bioinformatic model to enable the discovery of antibacterial peptides against multiple gram-positive pathogens. J. Chem. Inf. Model. 2016, 56, 588–598.
- Bayer, A.; Lammel, J.; Tohidnezhad, M.; Lippross, S.; Behrendt, P.; Klüter, T.; Pufe, T.; Cremer, J.; Jahr, H.; Rademacher, F.; et al. The antimicrobial peptide human beta-defensin-3 is induced by platelet-released growth factors in primary keratinocytes. Mediat. Inflamm. 2017, 2017.
- Juretić, D.; Vukičević, D.; Tossi, A. Tools for designing amphipathic helical antimicrobial peptides. In Antimicrobial Peptides; Springer: Cham, Switzerland, 2017; pp. 23–34.
- Seyfi, R.; Kahaki, F.A.; Ebrahimi, T.; Montazersaheb, S.; Eyvazi, S.; Babaeipour, V.; Tarhriz, V. Antimicrobial peptides (AMPs): Roles, functions and mechanism of action. Int. J. Pept. Res. Ther. 2019, 1451–1463.
- Wang, C.-K.; Shih, L.-Y.; Chang, K.Y. Large-scale analysis of antimicrobial activities in relation to amphipathicity and charge reveals novel characterization of antimicrobial peptides. Molecules 2017, 22, 2037.
- Chew, M.-F.; Poh, K.-S.; Poh, C.-L. Peptides as therapeutic agents for dengue virus. Int. J. Med. Sci. 2017, 14, 1342–1359.
- Sadredinamin, M.; Mehrnejad, F.; Hosseini, P.; Doustdar, F. Antimicrobial Peptides (AMPs). Nov. Biomed. 2016, 4, 70–76.
- Da Mata, É.C.G.; Mourão, C.B.F.; Rangel, M.; Schwartz, E.F. Antiviral activity of animal venom peptides and related compounds. J. Venom. Anim. Toxins Incl. Trop. Dis. 2017, 23, 3.
- Rautenbach, M.; Troskie, A.M.; Vosloo, J.A. Antifungal peptides: To be or not to be membrane active. Biochimie 2016, 130, 132–145.
- Faruck, M.O.; Yusof, F.; Chowdhury, S. An overview of antifungal peptides derived from insect. Peptides 2016, 80, 80–88.
- Muhialdin, B.J.; Hassan, Z.; Bakar, F.A.; Saari, N. Identification of antifungal peptides produced by Lactobacillus plantarum IS10 grown in the MRS broth. Food Control 2016, 59, 27–30.
- Mor, A. Multifunctional host defense peptides: Antiparasitic activities. FEBS J. 2009, 276, 6474–6482.
- Lacerda, A.F.; Pelegrini, P.B.; de Oliveira, D.M.; Vasconcelos, É.A.; Grossi-de-Sá, M.F. Anti-parasitic Peptides from Arthropods and their Application in Drug Therapy. Front. Microbiol. 2016, 7.
- Pretzel, J.; Mohring, F.; Rahlfs, S.; Becker, K. Antiparasitic Peptides. In Advances in Biochemical Engineering/Biotechnology; Springer: Berlin/Heidelberg, Germany, 2013; pp. 157–192.

