 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Michael Wolfe | + 1036 word(s) | 1036 | 2021-01-25 04:14:40 | | | |
| 2 | Camila Xu | Meta information modification | 1036 | 2021-03-30 05:29:55 | | |
Video Upload Options
γ-Secretase is an aspartyl protease.
1. γ-Secretase Inhibitors: Therapeutic Potential
The search for γ-secretase inhibitors (GSIs) as potential therapeutics for AD had gone on for over two decades, ever since Aβ was discovered to be a normally secreted peptide produced from a variety of cell types in culture [1]. Such inhibitors were identified even before the components of the protease were known and ultimately served as critical tools for discovery of presenilin as the catalytic component [2][3], as described earlier. Initially, these inhibitors were simple peptidomimetics (e.g., TSAs), but pharmaceutical companies quickly developed compounds with much better drug-like properties that allowed in vivo testing for the ability to lower Aβ in the brains of transgenic AD mice (e.g., expressing FAD-mutant APP and presenilin).
Acute treatment with GSIs did show such proof of principle for these drug candidates [4][5]; however, chronic treatment revealed serious peripheral toxicities [6][7], such as gastrointestinal bleeding, immunosuppression, and skin lesions, all effects that could be traced to inhibition of Notch proteolysis and signaling. As AD patients would be required to take GSIs for years and perhaps decades, the severe toxic consequences of γ-secretase inhibition caused great concern. While there were hopes for a therapeutic window that would allow lowering brain Aβ levels without the peripheral Notch-deficient toxicity, the failure of one GSI, semagacestat (Figure 1), in phase III clinical trials, dashed these hopes [8]. The trial resulted in unacceptable peripheral toxicities and—more worrisome—cognition that was worse than the placebo control groups.
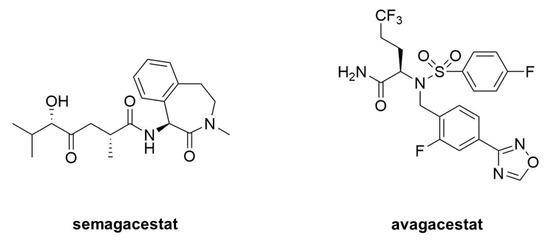
Figure 1. Clinical candidate γ-secretase inhibitors (GSIs). Semagacestat (left) is nonselective for APP vis-à-vis Notch, while avagacestat (right) is reported as selective for blocking γ-secretase proteolysis of APP over Notch.
Because all the serious toxic effects were apparently caused by inhibition of Notch signaling, the focus then went toward finding GSIs that could selectively inhibit the proteolysis of APP by γ-secretase without affecting Notch1 proteolysis. This led to the discovery of so-called “Notch-sparing” GSIs [9][10][11][12], a misleading term, as these compounds show APP/Notch selectivity and not complete lack of effect on Notch proteolysis. Moreover, the degree of selectivity was a matter of debate, with some reports of a lack of any selectivity for APP [13][14]. One such compound, avagacestat (Figure 9), went as far as phase II clinical trials, and like semagacestat caused Notch-deficient toxicities at higher doses, equivocal Aβ lowering in cerebrospinal fluid at lower doses, and worsening of cognition [15]. Evidence from mouse models suggest that the cognitive worsening may be due to increased γ-secretase substrates [16], although elevation of total Aβ, seen in plasma at low inhibitor concentrations, may be responsible [17][18]. These findings have effectively halted further development of GSIs for AD. Interestingly though, these compounds may be repurposed for oncology, for the treatment of various cancers that involve overactive Notch signaling [19].
2. γ-Secretase Modulators: Therapeutic Potential
While GSIs are out of further consideration for AD therapeutics, γ-secretase modulators (GSMs) are still of keen interest [20]. These compounds (see Figure 2 for examples) have the effect of lowering Aβ42 levels without decreasing overall Aβ levels or otherwise inhibiting general γ-secretase activity [21][22]. The decrease in Aβ42 is correlated with an increase in Aβ38, thereby replacing a highly aggregation-prone form of Aβ with a much more soluble form. Thus, these compounds can prevent the formation of plaques and other higher-order assembly states of Aβ42 in the brain. GSMs, however, have no effect, even at very high concentrations, on Notch proteolysis and signaling, nor do they elevate γ-secretase substrates. Presumably for these reasons, these compounds have shown excellent safety profiles, both in animal models and in human trials.
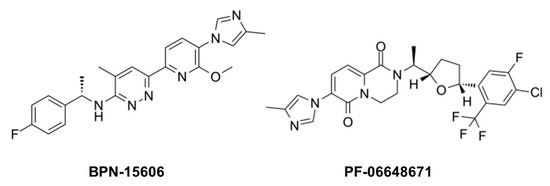
Figure 2. Example γ-secretase modulators (GSMs).
The mechanism of action of these compounds is not entirely clear, although the correlation between Aβ42 lowering and Aβ38 elevation is relevant, as Aβ42 is a precursor to Aβ38. γ-Secretase cleaves the Aβ42 C-terminus to release a tetrapeptide [23], and isolated γ-secretase converts synthetic Aβ42 to Aβ38 with release of this tetrapeptide [24]. Moreover, presenilin mutations decrease the Aβ42-to-Aβ38 conversion while GSMs stimulate it. Thus, GSMs appear to decrease Aβ42 by enhancing the carboxypeptidase activity of γ-secretase that converts this aggregation-prone peptide to Aβ38.
A critical issue with GSMs, however, like all anti-Aβ therapeutic strategies, is the design of clinical trials [25]. So far, all reported clinical trials with candidate AD therapeutic agents, including GSIs, GSMs and anti-Aβ immunotherapy, have been with individuals who already have AD or a pre-AD condition called mild cognitive impairment (MCI). Even in those with MCI, substantial neurodegeneration has occurred, and there are serious concerns that targeting Aβ after the onset of symptoms is too late. Aβ pathology in the brain may appear more than 10 years before the clinical manifestation of AD [26]. As Aβ pathology apparently precedes tau pathology [27][28], and tau pathology may then propagate from neuron to neuron [29][30], blocking Aβ after tau pathology is initiated may not prevent or slow the progression of AD and may not even prevent or delay disease onset. For anti-Aβ strategies—including GSMs—to succeed, clearer knowledge of the pathogenic process and timing is needed, as are convenient and reliable biomarkers and diagnostics.
Moreover, the clinical success of GSMs is completely dependent on whether Aβ42 is indeed the pathogenic entity in AD. The reasons for the focus on Aβ42 are arguably more historic than a result of an objective search with no preconceptions. Over 100 years ago, Alois Alzheimer described extraneuronal amyloid plaques as a signature pathological characteristic of the disease, and the discovery in the late 1980s and early 1990s that the primary protein component of the plaques is Aβ [31][32], with Aβ42 being the predominant species [33] and most aggregation-prone [34], led to the assumption that Aβ42 is the likely pathogenic species. Subsequent studies ranging from effects of APP and presenilin mutations to the neurotoxicity of various Aβ assemblies would seem to confirm this hypothesis. However, selectivity in the reporting of findings (negative results are more difficult to publish) in combination with incomplete knowledge of all forms of Aβ could result in mistaking correlation for causality. In light of more recent findings, mentioned earlier, that FAD mutations in PSEN1 increase Aβ peptide intermediates of 45 residues and longer, it would seem worthwhile to determine the effects of GSMs on the first or second trimming events of APP substrate by γ-secretase (e.g., Aβ49→46, Aβ45→42).
References
- Haass, C.; Schlossmacher, M.G.; Hung, A.Y.; Vigo-Pelfrey, C.; Mellon, A.; Ostaszewski, B.L.; Lieberburg, I.; Koo, E.H.; Schenk, D.; Teplow, D.B.; et al. Amyloid β-peptide is produced by cultured cells during normal metabolism. Nature 1992, 359, 322–325.
- Esler, W.P.; Kimberly, W.T.; Ostaszewski, B.L.; Diehl, T.S.; Moore, C.L.; Tsai, J.-Y.; Rahmati, T.; Xia, W.; Selkoe, D.J.; Wolfe, M.S. Transition-state analogue inhibitors of γ-secretase bind directly to presenilin-1. Nat. Cell Biol. 2000, 2, 428–434.
- Li, Y.M.; Xu, M.; Lai, M.T.; Huang, Q.; Castro, J.L.; DiMuzio-Mower, J.; Harrison, T.; Lellis, C.; Nadin, A.; Neduvelil, J.G.; et al. Photoactivated γ-secretase inhibitors directed to the active site covalently label presenilin 1. Nature 2000, 405, 689–694.
- Dovey, H.F.; John, V.; Anderson, J.P.; Chen, L.Z.; de Saint Andrieu, P.; Fang, L.Y.; Freedman, S.B.; Folmer, B.; Goldbach, E.; Holsztynska, E.J.; et al. Functional γ-secretase inhibitors reduce β-amyloid peptide levels in brain. J. Neurochem. 2001, 76, 173–181.
- Barten, D.M.; Guss, V.L.; Corsa, J.A.; Loo, A.T.; Hansel, S.B.; Zheng, M.; Munoz, B.; Srinivasan, K.; Wang, B.; Robertson, B.J.; et al. Dynamics of β-Amyloid Reductions in Brain, Cerebrospinal Fluid and Plasma of β-Amyloid Precursor Protein Transgenic Mice Treated with a γ-Secretase Inhibitor. J. Pharmacol. Exp. Ther. 2004, 27, 27.
- Searfoss, G.H.; Jordan, W.H.; Calligaro, D.O.; Galbreath, E.J.; Schirtzinger, L.M.; Berridge, B.R.; Gao, H.; Higgins, M.A.; May, P.C.; Ryan, T.P. Adipsin: A biomarker of gastrointestinal toxicity mediated by a functional γsecretase inhibitor. J. Biol. Chem. 2003, 278, 46107–46116.
- Wong, G.T.; Manfra, D.; Poulet, F.M.; Zhang, Q.; Josien, H.; Bara, T.; Engstrom, L.; Pinzon-Ortiz, M.; Fine, J.S.; Lee, H.J.; et al. Chronic treatment with the γ-secretase inhibitor LY-411,575 inhibits β-amyloid peptide production and alters lymphopoiesis and intestinal cell differentiation. J. Biol. Chem. 2004, 279, 12876–12882.
- Doody, R.S.; Raman, R.; Farlow, M.; Iwatsubo, T.; Vellas, B.; Joffe, S.; Kieburtz, K.; He, F.; Sun, X.; Thomas, R.G.; et al. A phase 3 trial of semagacestat for treatment of Alzheimer’s disease. N. Eng. J. Med. 2013, 369, 341–350.
- Netzer, W.J.; Dou, F.; Cai, D.; Veach, D.; Jean, S.; Li, Y.; Bornmann, W.G.; Clarkson, B.; Xu, H.; Greengard, P. Gleevec inhibits β-amyloid production but not Notch cleavage. Proc. Natl. Acad. Sci. USA 2003, 100, 12444–12449.
- Fraering, P.C.; Ye, W.; Lavoie, M.J.; Ostaszewski, B.L.; Selkoe, D.J.; Wolfe, M.S. γ-Secretase substrate selectivity can be modulated directly via interaction with a nucleotide binding site. J. Biol. Chem. 2005, 280, 41987–41996.
- Mayer, S.C.; Kreft, A.F.; Harrison, B.; Abou-Gharbia, M.; Antane, M.; Aschmies, S.; Atchison, K.; Chlenov, M.; Cole, D.C.; Comery, T.; et al. Discovery of begacestat, a Notch-1-sparing γ-secretase inhibitor for the treatment of Alzheimer’s disease. J. Med. Chem. 2008, 51, 7348–7351.
- Gillman, K.W.; Starrett, J.E.; Parker, M.F.; Xie, K.; Bronson, J.J.; Marcin, L.R.; McElhone, K.E.; Bergstrom, C.P.; Mate, R.A.; Williams, R.; et al. Discovery and Evaluation of BMS-708163, a Potent, Selective and Orally Bioavailable γ-Secretase Inhibitor. ACS Med. Chem. Lett. 2010, 1, 120–124.
- Crump, C.J.; Castro, S.V.; Wang, F.; Pozdnyakov, N.; Ballard, T.E.; Sisodia, S.S.; Bales, K.R.; Johnson, D.S.; Li, Y.M. BMS-708,163 targets presenilin and lacks notch-sparing activity. Biochemistry 2012, 51, 7209–7211.
- Chavez-Gutierrez, L.; Bammens, L.; Benilova, I.; Vandersteen, A.; Benurwar, M.; Borgers, M.; Lismont, S.; Zhou, L.; Van Cleynenbreugel, S.; Esselmann, H.; et al. The mechanism of γ-secretase dysfunction in familial Alzheimer disease. EMBO J. 2012, 31, 2261–2274.
- Coric, V.; van Dyck, C.H.; Salloway, S.; Andreasen, N.; Brody, M.; Richter, R.W.; Soininen, H.; Thein, S.; Shiovitz, T.; Pilcher, G.; et al. Safety and tolerability of the γ-secretase inhibitor avagacestat in a phase 2 study of mild to moderate Alzheimer disease. Arch. Neurol. 2012, 69, 1430–1440.
- Mitani, Y.; Yarimizu, J.; Saita, K.; Uchino, H.; Akashiba, H.; Shitaka, Y.; Ni, K.; Matsuoka, N. Differential Effects between γ-Secretase Inhibitors and Modulators on Cognitive Function in Amyloid Precursor Protein-Transgenic and Nontransgenic Mice. J. Neurosci. 2012, 32, 2037–2050.
- Lanz, T.A.; Karmilowicz, M.J.; Wood, K.M.; Pozdnyakov, N.; Du, P.; Piotrowski, M.A.; Brown, T.M.; Nolan, C.E.; Richter, K.E.; Finley, J.E.; et al. Concentration-dependent modulation of amyloid-β in vivo and in vitro using the γ-secretase inhibitor, LY-450139. J. Pharmacol. Exp. Ther. 2006, 319, 924–933.
- Barnwell, E.; Padmaraju, V.; Baranello, R.; Pacheco-Quinto, J.; Crosson, C.; Ablonczy, Z.; Eckman, E.; Eckman, C.B.; Ramakrishnan, V.; Greig, N.H.; et al. Evidence of a Novel Mechanism for Partial γ-Secretase Inhibition Induced Paradoxical Increase in Secreted Amyloid βProtein. PLoS ONE 2014, 9, e91531.
- Takebe, N.; Nguyen, D.; Yang, S.X. Targeting notch signaling pathway in cancer: Clinical development advances and challenges. Pharmacol. Ther. 2014, 141, 140–149.
- Bursavich, M.G.; Harrison, B.A.; Blain, J.F. γ-Secretase Modulators: New Alzheimer’s Drugs on the Horizon? J. Med. Chem. 2016, 59, 7389–7409.
- Weggen, S.; Eriksen, J.L.; Das, P.; Sagi, S.A.; Wang, R.; Pietrzik, C.U.; Findlay, K.A.; Smith, T.E.; Murphy, M.P.; Bulter, T.; et al. A subset of NSAIDs lower amyloidogenic Aβ42 independently of cyclooxygenase activity. Nature 2001, 414, 212–216.
- Golde, T.E.; Koo, E.H.; Felsenstein, K.M.; Osborne, B.A.; Miele, L. γ-Secretase inhibitors and modulators. Biochim. Biophys. Acta 2013, 1828, 2898–2907.
- Takami, M.; Nagashima, Y.; Sano, Y.; Ishihara, S.; Morishima-Kawashima, M.; Funamoto, S.; Ihara, Y. γ-Secretase: Successive tripeptide and tetrapeptide release from the transmembrane domain of β-carboxyl terminal fragment. J. Neurosci. 2009, 29, 13042–13052.
- Okochi, M.; Tagami, S.; Yanagida, K.; Takami, M.; Kodama, T.S.; Mori, K.; Nakayama, T.; Ihara, Y.; Takeda, M. γ-Secretase modulators and presenilin 1 mutants act differently on presenilin/γ-secretase function to cleave Aβ42 and Aβ43. Cell Rep. 2013, 3, 42–51.
- Golde, T.E.; Schneider, L.S.; Koo, E.H. Anti-Aβ therapeutics in Alzheimer’s disease: The need for a paradigm shift. Neuron 2011, 69, 203–213.
- Bateman, R.J.; Xiong, C.; Benzinger, T.L.; Fagan, A.M.; Goate, A.; Fox, N.C.; Marcus, D.S.; Cairns, N.J.; Xie, X.; Blazey, T.M.; et al. Clinical and biomarker changes in dominantly inherited Alzheimer’s disease. N. Eng. J. Med. 2012, 367, 795–804.
- Jack, C.R., Jr.; Knopman, D.S.; Jagust, W.J.; Shaw, L.M.; Aisen, P.S.; Weiner, M.W.; Petersen, R.C.; Trojanowski, J.Q. Hypothetical model of dynamic biomarkers of the Alzheimer’s pathological cascade. Lancet Neurol. 2010, 9, 119–128.
- Hardy, J.; Allsop, D. Amyloid deposition as the central event in the aetiology of Alzheimer’s disease. Trends Pharmacol. Sci. 1991, 12, 383–388.
- de Calignon, A.; Polydoro, M.; Suarez-Calvet, M.; William, C.; Adamowicz, D.H.; Kopeikina, K.J.; Pitstick, R.; Sahara, N.; Ashe, K.H.; Carlson, G.A.; et al. Propagation of tau pathology in a model of early Alzheimer’s disease. Neuron 2012, 73, 685–697.
- Liu, L.; Drouet, V.; Wu, J.W.; Witter, M.P.; Small, S.A.; Clelland, C.; Duff, K. Trans-synaptic spread of tau pathology in vivo. PLoS ONE 2012, 7, e31302.
- Glenner, G.G.; Wong, C.W. Alzheimer’s disease: Initial report of the purification and characterization of a novel cerebrovascular amyloid protein. Biochem. Biophys. Res. Comm. 1984, 120, 885–890.
- Masters, C.L.; Simms, G.; Weinman, N.A.; Multhaup, G.; McDonald, B.L.; Beyreuther, K. Amyloid plaque core protein in Alzheimer disease and Down syndrome. Proc. Natl. Acad. Sci. USA 1985, 82, 4245–4249.
- Iwatsubo, T.; Odaka, A.; Suzuki, N.; Mizusawa, H.; Nukina, N.; Ihara, Y. Visualization of Aβ42(43) and Aβ40 in senile plaques with end-specific Aβ monoclonals: Evidence that an initially deposited species is Aβ42(43). Neuron 1994, 13, 45–53.
- Jarrett, J.T.; Berger, E.P.; Lansbury, P.T., Jr. The carboxy terminus of the βamyloid protein is critical for the seeding of amyloid formation: Implications for the pathogenesis of Alzheimer’s disease. Biochemistry 1993, 32, 4693–4697.


